医薬品原薬の結晶多形スペクトルの読み方
本記載内容は、結晶多形の分析の専門家でない筆者が結晶多形のプロセス開発で得た経験、文献等から得た知識並びに専門家からの助言等を、まとめたものである。従って、記載した内容に不明な点、或いは間違った理解等からの内容があるかもしれない。ただ、本内容が結晶多形に遭遇した、或いはこれから知りたいと考えている方々の取っ掛かりの一助になれば幸いである。
医薬品原薬の結晶多形は1970年前後に知られており今まで多くの報告がされているので参照されたい。また、結晶多形の各種分析法の原理は各分析装置メーカーのホームページ並びにweb上で報文等をPDFで取得できるのでそれらを参照されたい(キーワード:結晶多形、化合物名、結晶多形の分析方法、IR、DSC、粉末 X 線回折、固体 NMR 及びそれらの組み合わせ)。
目次
初めに
1. 結晶多形(Polymorphism)とは?
2. 医薬品にとって結晶多形が重要であることとは
3. 結晶の性質と結晶形
4. 何故,同じ分子であっても結晶形が異なるのか?
5. 何故,同じ分子の結晶で物理化学的性質が異なるのか?
6. 何故,結晶多形が存在するのか?
7. 結晶多形の分子立体配座(コンフォメーション)の違いによる分子配列(パッキング)に充填様式
8. 結晶多形の分析法
9.
結晶多形の分析スペクトルの読み方
初めに
筆者は、治験薬(原薬)のプロセス開発中に結晶多形に遭遇したが、1995年当時は結晶多形について全く知らなかった。この時、結晶多形は特許が取得出来、原薬に関する特許が延長出来れば医薬品の独占販売期間が延長できることから、結晶多形の作製と勉強を始めた。
その時、結晶多形とは何? 何故、同じ原薬(分子)なのに溶解度が違うのか?結晶は最安定構造を取っていると思っていたのに、同じ化合物で数種類の結晶多形が存在するのか?等の疑問が湧いた。このことから、社内の結晶多形を研究していた方々にこれらの疑問と、何故、結晶多形は物理化学的性質が異なるのか? 結晶多形の見分ける方法(分析法)は? 各種分析スペクトルからどの様な情報が得られるか?等の質問をしたが、回答に最初はピンとこなかった。しかしながら、実際に結晶多形に遭遇しその作製と各種分析機器から得られるスペクトルを見ているとスペクトルの見方が少しずつ解って来た。すると何故、結晶多形は各種分析機器で得られる分析スペクトル上で違いが出るのか?の疑問が湧いた。その違いについて調べて行くと、結晶多形は結晶中の分子のコンフォメーション(立体配座)及び/或いは分子のパッキング(配列)が異なっていることが分かった。結晶中の分子のコンフォメーションは溶媒の種類(溶媒の物性)、溶解液の濃度、圧等により決定される。分子のパッキングは分子間及び/或いは分子(溶質)と溶媒間の水素結合、その他の分子間相互作用、並びに芳香環を有する化合物は分子(芳香環)間同士の
p-p
及び/或いはCH-p
相互作用、等で決まると理解するに至った。このことから、結晶多形は結晶中の分子コンフォメーション、分子パッキング並びに分子間相互作用等の違いが物理化学的性質を決めていると理解できた。次に、結晶多形の分析は、結晶状態で分析できる各種分析機器なら出来、分光法であるIR(ラマン)及び固体13C-NMR、熱分析法であるDSC(又は、TG-DTA)、立体構造解析法である
X 線回折(単結晶、粉末XRD)等により行える。結晶多形の判別は各種分析装置から得られたスペクトルの吸収ピークパターンとピーク強度を比較すると容易に判別(同定:定性分析)出来る。プロセス研究者は、各スペクトルから詳細な情報を得て他者に説明するためにも、分析機器の原理、スペクトル上に差異が出る理由などを知っておくべきである。また、結晶多形のスペクトルを解析出来るようになれば、分析中に何が起こっているのか、分析が正しい結果を示しているのか、得られた結晶形が単品か混合物か、並びに安定晶か不安定晶か等が判定できる。結晶多形の混合物の定量分析も出来るようになる。
また、結晶多形は、すでに物質そのものが公知であっても、
(i) 結晶形が新規であり、
(ii) 現行結晶形に比して有益な性質を有する場合(例えば、安定であること、体内吸収が良い等)は新規性がある発明として特許取得が可能となるため、医薬品原薬の開発にとって非常に重要である。
今回、医薬品原薬のプロセス研究(開発)者が結晶多形に遭遇した時に、結晶形の見分け方について最低知っておくべき各種分析法の原理と各種スペクトルの読み方について概要を説明したい。
既に、結晶多形の作り分けついては、「医薬品原薬のプロセス開発に於ける晶析のスケールアップ」で紹介しているので、参照されたい。
1. 結晶多形(Polymorphism)とは?
同じ分子(分子)の結晶であっても、
・結晶の安定性(結晶形転移)が異なる
・融点が異なる(図 1)
・結晶の形・色(外観)が異なる(図 2)
・血中濃度(バイオアベイラビリティ)が異なる(図 3)
・溶解度・溶解速度が異なる(図 4)
・結晶多形は、物理化学的性質が異なるため、固体で分析できる IR (Raman)、熱分析(DSC:示差走査熱量, DTA:示差熱分析)、粉末 X 線回折(Powder XRD)及び固体 13C-NMR で分析出来る
・結晶多形は、熱、圧力、溶媒等の媒介により結晶形転移(不安定晶から安定晶へ)する
等の異なった物理化学的性質を示し、その中でも結晶の安定性とバイオアベイラビリティは原薬の重要な品質に係わっている。
図 1 に示す様に、同じ化合物(分子)であっても物理化学的性質の違った結晶形が存在する。これらは結晶多形と呼ばれ、異なった融点と溶解度等を持っている。治験薬 A は最低 3 つの結晶多形(安定晶、不安定晶及び準安定晶)を持っている 1)(図 1)。結晶は融解する時熱吸収を起こし、結晶化する時発熱する。熱分析(DSC 或いは DTA)は、この性質を利用して熱分析中に結晶多形の融点と結晶化熱(熱媒介転移)が測定でき、結晶多形の区別、不安定晶から安定晶への転移も確認することが出来る。DSC 分析(スペクトル)から、治験薬 A の Type
A は不安定晶、Type B は不安定晶、Type C は準安定晶と判断できる。更に、結晶多形は、溶解度・溶解速度が異なるために、薬物(原薬)の薬効を示す血中濃度を確保できず原薬の効き目に差が出る。治験薬 A の不安定晶(Type A)と準安定晶(Type C)の体内吸収は不安定晶の方が良く、不安定晶の最高血中濃度は準安定晶より 2 倍高い値を示している(図 1)。
図 1. ある治験薬 A の結晶多形(Type A, B and C)のDSC スペクトルと Type A と Type C の血中濃度1)

1) 特許 WO2002020467より引用
結晶多形は物理化学的性質が異なることから、N-3,5-di-tert-buthylsalicylidene-3-carboxyaniline (1) の結晶多形は外観である色、形状が異なっている(図 2)2)。但し、結晶多形の判別は外観の違いだけで行うのは危険である。
図 2. N-3,5-di-tert-buthylsalicylidene-3-carboxyaniline
(1) の結晶多形2)

2) 上本紘平ら、「1A04 ホトクロミック化合物サリチリデンアニリン誘導体の 3 種類の多形結晶とその物性」より引用
何故、このようなことが起こるのか?化合物 1 の単結晶構造解析から、図 3 に示す様に、化合物 1 の外観(色・形状)の違いは、ベンゼン環間の二面角、分子コンフォメーションの違いによる共役系の電子の流れに差が生じるためと考えられる。

金庭らは3)、インドメタシン (2) の結晶多形 a(I)* form と b(II) form を第十改正日本薬局方の溶出試験器を使用し、緩衝液(日本薬局方,第2液)を用い緩衝液の温度を変化させ平衡濃度法により溶解度を測定した結果を報告している(図3)。一般的に、溶解度は不安定晶が良く、安定晶は文字通り熱安定であるが良いため溶解度が悪い。インドメタシン (2) の a form は全ての温度で高い溶解度を示していることから、a form は不安定晶(準安定晶)と考えられる。
図 4. インドメタシ或いはン (2) の結晶多形の溶解速度と溶解度

医薬品原薬の結晶多形が重要であると認識された例として、解熱鎮痛薬であるアセチルサリチル酸(アスピリン) (3) が有名である。アスピリンはバイエル社によってヤナギの枝などにふくまれるサルチル酸から開発された医薬品であるが、他の会社でも次々と製造された。しかしながら、バイエルのアスピリンは他社のものよりよく効くという噂が流れた。その噂は事実であり、後日、同じ化学構造(分子)をもつアスピリンでありながらバイエルのものは他社の結晶形と違っていたことが判明した。アスピリンの結晶形の違いが体内への吸収と効き目(効果)、胃への副作用にも差があることが理解された。三上らは4)、アスピリンの結晶多形を作製し、市販の結晶形は図 5 の D であることを示している(図 5)。
図
5. アスピリン
(3) の結晶多形

4) 三上貴司、新潟大学工学部化学システム工学科化学工学コース晶析工学研究室より
医薬品原薬の化学構造(分子)が同じであっても、結晶形によって体内での溶解速度が異なり体内への吸収に差が出るため血中濃度に差を生じ、薬効に違いが生じる。結晶多形は医薬品原薬の品質に重要な試験項目であり、その性質(物性、安定性、多形転移)、分析方法並びに製剤研究に於いてその重要性が認識されている。下記の図 6に示した様に、渡辺らは5)各種再結晶溶媒から得られたアスピリンの血中濃度を示している。商用アスピリンより水から再結晶した結晶形は早い吸収と高い血中濃度を示している。
図
6. アスピリン
(3) の各種再結晶溶媒による結晶癖と結晶多形の溶解速度

この様に、医薬品原薬の結晶形の違いは溶解度の違い、吸収速度の違い並びに血中濃度の違いとして現れ薬物の有効性に差が出る。これは医薬品にとって致命的である。このことから、医薬品原薬の結晶多形の同定と判別は非常に重要である。
特許に従えば1)、化合物 5 は3種類の結晶多形が確認されている。特に、DSC 分析からは Type A が不安定晶、 Type B が安定晶及び Type C が準安定晶として帰属している(図 7)。結晶多形の各分析機器から得られるスペクトルを重ね合わせることにより吸収ピークの強度とパターンの違いが一目で区別と同定が出来る(図 7 及び 8)。
図 7. 化合物
5 の結晶多形(Type
A, B and C)の熱分析(DSC)とFT-IRスペクトル


図
8. 化合物
5 の結晶多形(Type
A, B and C)の粉末 X 線回折スペクトル

医薬品原薬の個体には、結晶とアモルファス(無定形結晶)が存在する。原則的に、同じ分子が規則正しく配列し固体を形成したものを結晶と呼んでいる。同じ分子であっても不規則に配列した個体(粉体)をアモルファスと呼んでいる。同じ分子の結晶であっても物理化学的性質並びに/或いは外観が異なる結晶、或いは外観が異なるが同じ結晶形である結晶も存在する。これは結晶癖と呼ばれ、同じ分子の水和物・溶媒和物等は疑似結晶多形と呼ばれる(図
9)。

・結晶多形とは、同一分子でありながら分子のコンフォメーション或いは/並びに分子の配列(パッキング)等が異なり分子間の相互作用等が違うことにより物理化学的性質の違いを示す結晶である。
・疑似結晶多形とは、同じ分子の水和物或いは溶媒和物の結晶のことを言う。疑似結晶多形は加熱して行くと水分或いは溶媒が除去され、除去された溶媒等が存在していた位置が空間となったり、その空間を埋めるために結晶形転移を起こすことがある。基本的には、結晶多形として取り扱わない。
・結晶癖(外観)とは、結晶形は同じであっても結晶の成長方向が違うため、結晶の外観が異なる結晶のことである。これらの結晶は外観だけが異なるだけであり、物理化学的性質同じである。これは結晶多形として取り扱わない。
結晶形の違い(結晶多形)は、同一分してあっても結晶中の分子のコンフォメーション(立体配座)及び/或いは分子のパッキングの違により発生する。このようにして生じた結晶は同じ分子でありながら物理化学的性質が異なることから結晶多形と呼ばれている。殆どの場合は、結晶多形は異なった外観(色、形)並びに熱安定性を有することが多い。
(1) 同一分子であっても結晶中の分子のコンフォメーション、分子のパッキングの違いは、分子間の水素結合、π-π並びに/或いは CH-p 結合の様な分子間相互作用の強度の異なり、結晶の物理化学的性質の違い及び結晶の硬さ等として現れる。この違いが結晶多形の硬さ、分子間相互作用の強さ、融点、溶解速度、溶解度、生物学利用能(バイオアベイラビリティ)並びに熱安定性等を左右させ、医薬品原薬の品質に重大な影響を与える。また、結晶多形では外観(色と形)が異なる場合がある。
5-Methyl-2-[(2-nitrophenyl)amino]-3-thiophencarbonitrile
(6) の結晶群は外観(形並びに色)が異なるが、同一分子の結晶である。これらは結晶多形である。結晶内の分子コンフォメーション及び配列(パッキング)が異なることから、結晶癖並びに物理的性質に差を生じさせ結晶の外観に違いを発生させている(図 10)。
図
10. 5-Methyl-2-[(2-nitrophenyl)amino]-3-thiophencarbonitrile
(6) の結晶多形と結晶の外観7)

7) S. R. Byrn, et al., J. Amer. Chem.
Soc., 122(4), 585-591 (2000) より引用
(2) アモルファス(無定形結晶)
全ての個体(粉末含む)が結晶を形成していることはなく、急激な晶析法で析出させると分子がバラバラに凝集し、個体として析出する場合がある。これがアモルファス(無定形結晶)である。アモルファスは、溶解性及びバイオアベイラビリティ等が良いが、結晶でないため分子間相互作用が弱く安定性が悪い場合が多い。アモルファスは安定性が悪いため医薬品原薬に採用されことが多くないが、後発品(ジェネリック)では結晶形の特許を回避するためにアモルファス(粉末)を採用する場合がある。
アモルファスとは、
・粉末 X 線回折分析では、乱反射するため結晶の様にピークを与えことはない
原薬の品質は純度・不純物量・不純物プロファイル・外観(色・粒子径)等が有効性と副作用に重要な影響を与える。結晶の品質は製剤化・有効性に重要な役割を担っており、特に、結晶多形は溶解度、溶解速度、安定性並びにバイオアベイラビリティ(血中濃度)を左右する重要なファクターである。
同じ分子であっても結晶形が異なるのは、結晶中の分子の立体配座(コンフォメーション)と配列(パッキング)の違いにより分子間の相互作用の差がある結晶が発生するためである。
1) 溶液中の分子は自由に運動しているが、濃縮或いは冷却により溶解度が低下すると飽和状態となり、分子は溶媒との相互作用等による安定コンフォメーションを取り、更に分子間相互作用(水素結合、p-p interaction、C-H-p interaction 等)が強くなり、分子が集合してくる。更に過飽和状態が高くなると分子が集合しクラスターを形成し、クラスターが凝集して核が形成され、核に分子或いはクラスターが集合・凝集して結晶となって系外(結晶化)へ出される。この様にして、分子のコンフォメーション或いは/及び分子配列(パッキング)の違いによる結晶が形成される。これが結晶多形である。結晶多形は同じ分子の結晶であっても分子コンフォメーション及び/或いは分子パッキングの違いが分子間相互作用の違いとなる。
但し、分子はある条件下での安定な分子コンフォメーションを取っている場合と数分子集合して安定化してコンフォメーションを取っている場合がある。
一般的な分子間相互作用とN-3,5,-di-tert-buthylsalicylidene-3-carboxyaniline (1) の結晶多形の分子立体構造(コンフォメーション)を図 11 に示す。これらの相互作用等の違いが結晶の外観に違いを発生させている。
図
11. 分子間相互作用と分子コンフォメーション
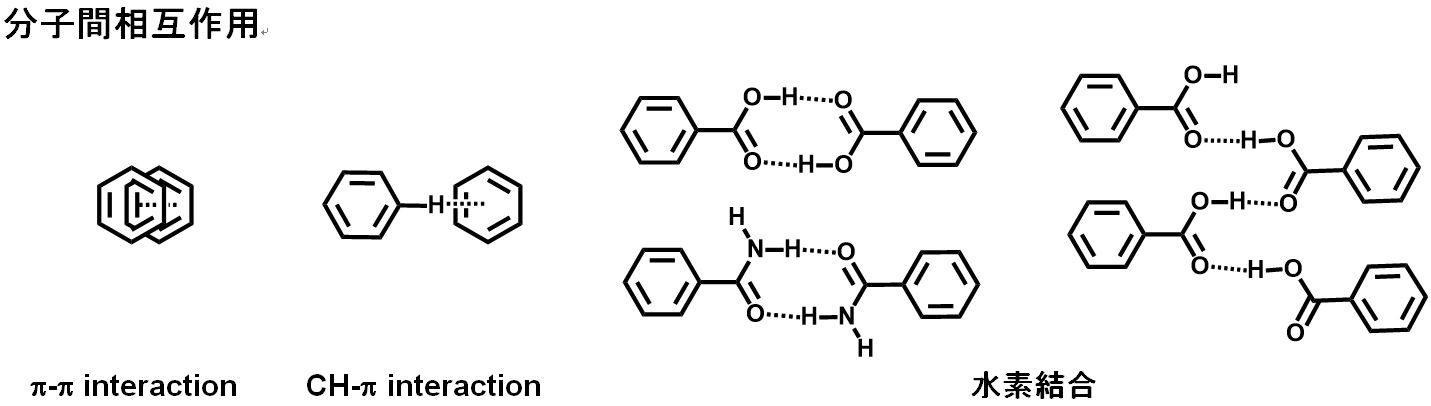

8) 豊橋技科大工 小畑ら、結晶計算法によるアスピリン結晶の配座多形解析より
何故、結晶多形が物理的性質に差を生じさせるのか?結晶多形をどの様に分析すればよいのか?の疑問が湧いてくる。結晶のクラスターや核が形成される際に、分子自身の極性、ファンデルワールス力、分子間水素結合等による分子間結合並びに溶液濃度、熱、圧力、溶媒種等との相互作用(水素結合、溶媒和等)の力が働き、溶媒に溶解した分子と溶媒との分子間相互作用や分子コンフォメーションが決まってくる。結晶多形はこの様に晶析条件(溶媒の種類、濃度、過飽和状態、冷却速度等)の違いで分子コンフォメーションと分子パッキングに違いが発生するために生じる。結晶中で分子のコンフォメーションとパッキング様式及び分子間相互作用の強度が違う結晶多形は、結晶の外観(色と形)、性質並びに熱安定性等の異なった物性(物理化学的性質)を示す。
例えば、アスピリン(図 12A)及びアセトアミノフェン(図 13B)のそれぞれの結晶多形(form I と form II)は分子間水素結合様式の違いと分子パッキングの違いを示している。
アスピリン (3) の 結晶多形は分子間水素結合の違いが分子パッキング(配列)の違いとなっており、form I は安定晶であり、form II は不安定晶である9)。
図 12A. アスピリン (3) の結晶分子間水素結合と分子パキング9)

図 12B に示したアセトアミノフェン (7)
の分子パッキングの違いは分子間水素結合の違い(或いは、分子間水素結合の違いが分子パッキングの違い)として現れる。結晶多形である form I は熱力学的に安定(安定晶)であり、form II は form I に比較し熱及び圧に対して熱力学的に不安定(不安定晶)である10)。Boldyrevaらは11)、アセトアミノフェンの結晶中の分子パッキング(配列)様式により熱安定性が影響され、熱安定性は結晶中の分子密度と反比例していることを示している。また、アセトアミノフェンの結晶多形である form III は準安定晶であり、特定の条件下でのみ存在する。
図 12B. P-アセトアノフェン (7) の結晶分子間水素結合と分子パッキング10)

9) M. Huremovic, et al., J. Chem. Bio. Phy. Sci. Sec. A, 7(1), 231-246 (2017).
10) K. A. Lyssenko, et al., Chem. Commun., 46, 3469–3471 (2010)
11) E. V. Boldyreva, et al., Phys. Chem. Chem. Phys., 13, 14243–14253
(2011)
アセトアミノフェン (7) の市販原薬は大半が結晶形 form I と言われている12)。アセトアミノフェン (7) の結晶多形の粉末 X 線回折スペクトル (XRD) を図 12C に示すが、各結晶形の XRDスペクトルはそれぞれ異なったピークパターンを与えることから容易に区別することが出来る。
図 12 C. アセトアミノフェン (7) の結晶多形のXRD スペクトル12)

また、図 13 に示す様に、大橋は2)、N-3,5,-di-tert-buthylsalicylidene-3-carboxyaniline (1) には a、b及び g 晶の 3 個の結晶多形が存在することを報告している。それぞれの結晶多形は分子中の 2-aminobenzoic acid の回転による芳香環同士の平面角の違いが生じている。この分子コンフォメーションの違いが、分子間相互作用の強さの違いとして現れ、結晶の外観(形状と色)に影響を与えている。
図 13. N-3,5-di-tert-buthylsalicylidene-3-carboxyaniline
(1) の結晶多形の分子コンフォメーション

2) 東工大理工 大橋、「ホトクロミック化合物サリチリデンアニリン誘導体の 3 個の多形結晶とその物性」より引用
以上の様に、結晶多形は、結晶中の分子コンフォメーション及び分子パンキング並びに分子間水素結合等の違いが分子間相互作用力の違いを与え、結晶の物理化学的性質(外観、溶解度、溶解速度、熱安定性等)の違いとして現れる。結晶多形のこの違いが固体状態で分析できる分析機器で違いとして現れる。
既に、「5. 何故,同じ分子の結晶で物理化学的性質が異なるのか?」で述べた様に、同じ分子であっても溶解している分子は溶媒(水も含む)との相互作用の下で自由運動しているが、溶解液(溶媒)の濃縮或いは冷却等で溶解度が下がって行くと分子運動が制限され分子と溶媒との相互作用とその条件下での分子の安定コンフォメーションを維持しながら分子間の相互作用が強まる(図 15)。分子間相互作用が強まると分子が集合→凝集→核形成が起こり、核が成長して結晶が形成される。この時、分子と溶媒(溶解液)並びに分子間相互作用が温度と濃度等により違いにより溶液中の分子コンフォメーションを維持しながら分子の配列(パッキング)が決定されるために。結晶多形が形成される。
影近らは13)、N-Hydroxy-N-phenylbenzamide
(8) が溶媒種(溶媒の物性)により溶液中でオキシム基のN-OHとカルボニル基が cis 或いは trans 体のコンフォメーションを形成し、溶解度が下がっていくとその分子コンフォメーションを維持して結晶を与えることをNMR の NOE 測定から示している(図
14)。
図 14. N-Hydroxy-N-phenylbenzamide
(8) の結晶多形の作り分け

13) H. Kagechika, et al., Crystal Growth & Design, 2006, 6
(9), pp 2007–2010より引用
また、大島らは14)、タルチレリン水和物(8)(効能・効果:脊髄小脳変性症における運動失調の改善)が溶媒種(CD3OD-D2O)によって分子のコンフォメーションに影響を受け、その影響を受けたまま結晶多形を作り分けられることを明らかにしている(図 15)。NMR分析に於いて、化合物 9 はD2O 中で NOE が観測されたが、CD3OD の比率が増えると共に NOE が観測されなくなっている。それらの結果から考えられる分子コンフォメーションと単結晶の X 線構造解析から得られた分子コンフォメーションとの間に相関があった。化合物
9 の結晶は CD3ODから
b
form晶が、D2O から
a
form 晶が得られている。
図 15. タルチレリン水和物 (9) の結晶多形

14) 大島 寛 「ナノ医薬製造と核形成」より引用
W. Wang らは15)、furosemide (10) の結晶多形を溶媒種の変更により作成し、各種溶媒から疑似多形を含め Form I ~ Form V を得ている。これらの結晶多形の同定は粉末 X 線回折により実施された(図 16)。
以上の様に、溶媒は、分子との相互作用を反映して分子コンフォメーションを規定しそのコンフォメーションを維持したままパッキングさせるため、結晶多形を作り分けることを示している。

15) W. Wang, et al., Bull.
Korean Chem. Soc., 30(10) 2265-2268 (2009)
7. 結晶多形の分子立体配座(コンフォメーション)の違いによる分子配列(パッキング)に充填様式
化合物 11
(V102862)は、図 17 に示す様に、分子コンフォメーションの違いが結晶への分子パッキングの違いを生じさせ、分子間の水素結合様式が異なる2種類の結晶多形を与える16)。この分子のコンフォメーション及びパッキングの違いは、分子間水素結合様式並びに分子間相互作用(静電結合等)に差を生じさせ、物性の違った2種類の結晶多形を与えている。
図 17. 化合物 11 の結晶分子パキング様式16)

16) XRPD Crystallographic analysis http://www.analytics-pharm.com/index.php/crystallographic-analyses より引用
同じ分子であっても結晶中の分子コンフォメーション(立体構造)・分子パッキング(配列・充填)様式が異なれば、結晶内の分子間の相互作用(水素結合様式、芳香環とhydrocarbonとのCH-p 並びに芳香環同士の π-π インターラクション等)等に差を生じさせ、性質の違う結晶が生じる。これが結晶多形である。結晶多形の分子の立体構造並びに分子間相互作用の異なりは、結晶分子中の各原子と置換基(芳香環、カルボニル基(C=O)、水酸基(OH)、水素(C-H)等)の距離・角度等の環境に差を生じさせる。この置換基・分子コンフォメーション・分子パッキング等の環境の違いは結晶多形として現れ特有な各種分析スペクトルを与える。
結晶中の分子コンフォメーション並びに分子パッキングの差は分子間相互作用(水素結合、π-π interaction、 C-H-π interaction、静電力並びに原子間結合角・結合距離等)に差を生じさせる。分子のコンフォメーション及びパッキングの違いは、IR(水素結合、原子間距離、二面角の違いにより原子間の伸縮振動・ベンディング・置換基の極性の差等)、粉末X線回折(分子の立体構造、分子のパッキング)、熱分析(DSC, DAT:融点、熱安定性と分解熱)等に差が出る。
結晶多形は、同じ分子の結晶が異なった分子コンフォメーション或いは分子間相互作用等が異なるために、物理化学的性質差を利用して分析機器で区別できる。
結晶多形を判別できる分析機器は、
・IR(Raman)
・粉末X線回折
・DSC(示差走査熱量測定:Differential scanning calorimetry)
・個体NMR spectrum
1) IR(赤外線吸収スペクトル)の測定原理の概要と代表的なスペクトル
物質(分子)は赤外線が照射されると光エネルギーを吸収して伸縮・回転する。この時、分子中の官能基は赤外線の特定の波長を吸収して伸縮・変角振動する(図 18)。結晶内の分子の充填様式及び立体構造が異なれば、分子中の原子間の距離並びに結合角の環境の差として現れる。赤外線を照射すると環境の違う分子の官能基は異なった特定の赤外線の波長を吸収するようになり、その吸収量(強度)に差を生じさせる。IRスペクトルはこの差を利用して結晶多形を見極めることが出来る(図 19)。
図
18. IR 吸収スペクトルに与える分子中の原子間伸縮及び変角振動17)

17) 中西香爾、「赤外線吸収スペクトル -定性と演習-」より引用
図 19. FT-IR(フーリエ変換赤外分光光度計)の原理18)

18) 赤尾賢一 九州大学中央分析センターニュース「フーリエ変換赤外分光光度計(FTIR)①」より引用
19) B.
Akahlaghhinia, et al., Turk. J. Chem.,
42、170-191(2018)より引用
インドメタシン (2) は各種溶媒から種々の結晶多形が得られているが、その判別にIR スペクトルが用いられることが多い。IR スペクトルは、吸収波長とその強度が異なっていることが多く、吸収強度とそのパターンから結晶多形を同定・定量することができる。一般的に、結晶多形を論議する場合は、図 20 に示す様に、IRの吸収スペクトルを構造解析とは逆に表す。
図
20. Indomethacine (2) の結晶多形の IR Spectrum20)

20) Bratu, et al., STUDIA
UNIVERSITATIS BABEŞ-BOLYAI, PHYSICA, SPECIAL ISSUE, 315 (2001)より引用
結晶多形は、医薬品原薬の粉砕時、或いは IR を測定時に KBr と試料を混ぜ均一サンプルを作製するために強く磨り潰すと圧(或いは熱)かけると転移(圧媒介転移、熱媒介転移)を起こす可能性があるのでサンプル調整時に注意を要する。
2) 粉末 X 線回折法(XRD : X-ray Diffraction)の測定原理の概要と代表的なスペクトル
粉末 X 線回折法は、単結晶の粉末集合体の様な多結晶試料にX線を照射(入射角)すると各回折角(2q)について回折強度を測定することにより粉末X線回折パターンを得、未知の結晶並びに結晶多形の同定、定量及び結晶化度並びに結晶多形の混合比等の測定などを行う方法である(図 21)21),22)。
図 21.
Bragg 回折とX線 回折装置の原理図21)

21) 神鳥和彦、「粉末X線回折測定による固体構造の研究」より引用
22) 日本薬局方より引用
粉末 X線回折は結晶多形の区別(同定(同定)、混合物の定量)、結晶と非結晶(アモルファス)の区別並びに結晶化度を測定するのに有用である。図 22-A は結晶多形の粉末 X 線回折(XRD)スペクトルであるが、試料(結晶多形)に入射X線が照射されると特有の回折角で回折 X 線が検知されピークを与える(結晶形 A~D)が、非結晶(アモルファス)では入射 X 線が乱反射するため特有の回折 X 線が見られないためピークを与えない20)。結晶多形が混合して結晶化された混合物、或いは人為的に結晶多形を混合した混合物は、図 22-B に示す様に、結晶多形の粉末 X線回折スペクトル a) 及び b)の回折ピークを重ね合わされたパター ン c)のスペクトルを与える21)。このことから、粉末 X線回折スペクトルから結晶多形の混合物であっても見極められる。
図 22. 結晶多形の粉末
X 線回折スペクトル22)

23) 深水、薬剤学, 65(4), 239-242
(2005)
結晶多形のスペクトルが結晶性化合物の粉末 X 線回折パターンは各化合物の各結晶形に固有であり特有であり、以下の情報を得ることが出来る(Table 2)。
Table 2. X
線回折パターンから得られる情報24)

結晶が大きく成長すると結晶子が大きく、結晶が成長せず小さいと結晶子が小さくなる(図23)。結晶子のサイズが XRD スペクトルに与える影響は結晶子が大きいと回折 X 線が散乱しなくなるため鋭いピークを与える。しかし、結晶子が小さい結晶が集合した結晶では、結晶子の並びが不規則となり回折 X 線が散乱するため幅広いピークを与える21)。
図 23. 結晶サイズと粉末 X 線回折スペクトル21)

結晶多形の XRD スペクトルは、測定試料の調製の仕方或いは試料の載せ方等により回折ピークが少しずれる或いはブロードになることがあるが、ピークのパターンは変わらない。従って、XRD で結晶多形を同定する場合は、スペクトルの全体のピークの高さ比が同じでピークパターンが同じであることを確認する。
3) DSC(示差走査熱量測定:Differential scanning calorimetry)の測定原理の概要と代表的なスペクトル
熱分析には、DSC(示差走査熱量測定)とDTA(Differential
thermal analysis:示差熱分析)測定装置の両方があるが、結晶多形の熱分析には DSC が用いられることが多い。
DSC は、物質および基準物質の温度をある一定の温度プログラムに従って変化させながら、その物質と基準物質に対するエネルギー入力の差を温度の関数として測定する技法である。試料の融解,ガラス転移,熱履歴,結晶化,硬化,熱変成などによる試料の温度変化を評価することが可能である。この温度差が単位時間当たりの熱エネルギーの入力差に比例するように設計されている点がDTAと異なっている(図 24)25)。しかし、結果として DTA
と DSCは同等のスペクトルを与え、同等に取り扱える。DSC は、結晶の融点とその後の相転移(融解・気化・結晶化・分解等)を連続的に測定でき、結晶の安定晶か、不安定晶かを判別できる。DTA と TG(Thermogravimetry;熱重量測定)は組み合わせられていることが多く、熱量と重量変化が同時に測定出来るために水和物或いは溶媒和物結晶の水分量或いは溶媒量等を分析できる。
図 24. 熱分析の原理と表示されるスペクトル25)

25) 日立ハイテク 大久保信明熱分析の基礎と応用より引用
DSCは熱量の吸収と発熱を測定することが出来るため、DSC スペクトル上に化合物の融点(溶融による熱吸収)、溶融後の液相から発熱を起こして結晶化(固体)へ相転移、更には分解により分解熱の発熱も観察できる。カルバマゼピン (12)26)は、174.4℃ 付近で溶融が始まり 176.2℃ で完全な融解(融点)し、178℃で結晶化熱(発熱;不安定晶から安定晶へ転移)により固体(結晶)へ相転移している。従って、カルバマゼピンの結晶はDSC スペクトルから不安定晶であると考えられる(図 25)。
図 25. カルバマゼピン
(12) の
DSC スペクトル26)

結晶多形は、 DSC測定時に昇温速度が速いと結晶化熱(発熱ピーク)を測定できない、或いは融点が近い場合は単一融点を与える場合があるので、昇温速度に注意を要する。
4) 固体
13C-NMRの測定原理の概要と代表的なスペクトル
固体13C-NMRでは、スペクトルの違い、緩和時間の違いから結晶多形の判別は可能。13C、15N、23Na、31Pなどによる測定・評価が可能である。一般的にCP (Cross Polarization) / MAS(Magic Angle
(54.74o) Spinning)法によりスペクトルが測定できる。
固体 13C-NMR は、試料の入ったプローブを静止状態で測定すると図 26 の(a)の様に分子の運動が抑えられているため各スピン間の時期的相互作用が平均化できないためシグナルの線幅は広くなるが、プロトンデカップリング下で測定するとプロトンの影響が除かれる(b)。固体試料の入ったプローブをマジックアングル 54.74° に倒し(Aの状態)高速回転させると溶液中の高速回転が磁気的相互作用を平均化しているのと同じ効果を生むため炭素が分離したシグナルを与える(C)25)。
図 26.
個体C13-NMR のスペクトルと原理27)

チアミン塩酸塩 (13) には 2 種類の結晶多形が確認されているが、結晶多形の I 形晶とII 形晶は個体 C13-NMR スペクトル上に於いて、幾つかの炭素のケミカルシフトが異なっている28)。このことは、結晶中の分子のコンフォメーション或いは/及び分子パッキングが異なっていることを意味しており、結晶多形はC13-NMR で判定することが可能である(図 27)。
図
27. チアミン塩酸塩
(13) の個体 C13-NMR のスペクトル

9.
結晶多形の分析スペクトルの読み方
今まで述べてきた様に、結晶は結晶中の分子のコンフォメーションと分子のパッキングの仕方により分子間水素結合、並びにその他の分子間相互作用の強さが異なる。この相互作用の強さの差が結晶多形の物性(物理化学的性質)の差となる。従って、結晶多形の判別は、結晶の状態で分析できる 分光方(IR、ラマン、固体NMR 等), 熱分析法(DSC, TG-DAT等)並びにX 線回折法(単結晶、粉末)の各種分析装置から得られるスペクトル上に違いとして現れる。
Kaneniwaらは29)、図 28 に示す様に、Chloramphenicol palmitate (4) の 3 種類の結晶多形(A 形晶: 安定晶、B形晶: 準安定晶及び C 形晶: 不安定晶)について、粉末 X 線回折(powder XRD)、IR 及び DSC
の各種分析機器を用いて分析している。
図 28. Chloramphenicol palmitate (4) の各種分析機器のスペクトル29)

化合物 4 の 3種類の結晶多形は powder XRD、IR 及び DSC から得られたスペクトルのピークパターンにより結晶形 (a) A 形晶、(b) B形晶及び (c) C形晶の違いを容易に判別・同定できる。 化合物 4 の DSC スペクトルからA 形晶は
95℃ 付近に融点と考えられる鋭い熱吸収ピーク
があり安定晶と考えられる。B形晶
とC形晶
は
90℃ 付近に同じ融点を持っているが、C形晶は 72℃ 付近に発熱ピークが観測されている。この発熱ピークは、スペクトル上に熱吸収ピークが観測されていないが、結晶化熱と考えられる。DSC 分析条件である昇温速度が速いためか或いは本結晶の性質かは分からないが、C形晶は融点の熱吸収ピークが観測されずに結晶が熱媒介転移を起こしたものと考えられる。従って、各結晶多形の融点から、A 形晶は安定晶、B形晶は準安定晶、C形晶は不安定晶と判断できる。また、これらの結晶多形は
C形晶(不安定晶) から
B形晶(準安定晶)へ転移を起こすが、C形晶及び
B形晶から A形晶への熱媒介転移は起こらないものと考えられる(図 29)。
29) N. kaneniwa, et
al., Chem. Pharm. Bull., 33(4)
1660-1668 (1985) より引用
1) IR (Raman) スペクトルの読み方
結晶多形は結晶中の分子のコンフォメーション、分子のパッキング、水素結合等の分子間相互作用がことなるため、原子間距離並びに原子間結合角が異なる。このことから、結晶に外部から赤外線(或いは Raman 光)が照射されると特定の波長を吸収し分子内で特有の原子間運動が起こり、結晶特有の吸収波長及び強度に違うスペクトルを与える。結晶多形の見分け方は、例えば、インドメタシン (2) は、少なくとも 4種類の結晶多形があり、図 29 の IR
スペクトル上の で示した 1700 cm-1(C=O)付近、1500~1400 cm-1付近、1300~1100cm-1付近及び 900 cm-1~(指紋領域)で吸収波形と強度の違いを示している。このことから、IR スペクトルは結晶多形を同定(判定)することが出来る30)。また、IRスペクトルは結晶多形の混合物の同定(定性)並びにその混合比の定量も可能である(図 29)。
結晶多形の IR スペクトルの表し方は機器分析で示す吸収方向とは逆に表すのが一般的であ
る。
る。
図 29. インドメタシン (2) の結晶多形の
IR スペクトルの比較30)

30) Bratu, et al., STUDIA UNIVERSITATIS
BABEŞ-BOLYAI, PHYSICA, SPECIAL ISSUE, 315 (2001)より引用
図 30. Mebendazole (14) の結晶多形 A
形晶、B 形晶及び C 形晶のIR
吸収スペクトル31)

31) H. Y Aboul-Enein, et al., Spectroscopy Letters, 34(5), 527-536
(2001)より引用
Aprepitant (15) には、form I と form
IIの結晶多形が知られており、図 31 の a) に示す様に、非常に似た IR スペクトルを与えるが、結晶多形の form Iは非常に小さいが1140 cm-1 付近に特有のピークを持っている32)。 b) に示す様に、Aprepitant の IR 吸収スペクトルは、form Iに form IIを添加量と吸収強度から検量線を作成することにより、混合した結晶多形の未知試料の混合比を定量出来ことを示している。
図 31. Aprepitant (15) の IR 吸収スペクトルから結晶多形の混合物比の定量32)

32) サーモフィッシャーサイエンティフィック株式会社 Application Note
M12006 より引用
2) 粉末 X 線回折(powder
XRD)の読み方
粉末 X 線回折測定で分かることは、ピークの有無で結晶か非結晶か、ピークの位置と強度で同定(定性分析)、結晶多形の特有のピークの高さから混合物の定量分析、ピークの半値幅で格子の歪みと結晶子サイズ、並びにピークの幅(潰れたピーク)による結晶化度等である(図 33)33)。
同じ結晶試料でも、サンプルの調整によりスペクトル上のピークが少し位置(回折(2q)角のピーク)のズレ、或いはピークがブロードになることがある。
図 32. 粉末 X 線回折(powder XRD)スペクトルで分かること21),33)

33) 中山将伸のHP 、https://mmnakayama.jimdofree.com/study/粉末x線回折%EF%BC%91/より引用
結晶と非結晶(アモルファス)のXRD スペクトルは、結晶(結晶形 A)では回折角(2q/deg.)での反射強度に従いピークがそれぞれ強度と分離して観測されるが、非結晶(アモルファス)はピークを与えないが、結晶と非結晶の混合物は図 33 で示す様に混合した化合物の XRD スペクトルを与える。結晶化度の計算は結晶の回折 X 線ピーク強度とアモルファスのピーク強度を足した状態でスペクトルのピークを与えるため、結晶とアモルファスのそれぞれのピーク強度面積から実施できる34)。
図 33. 結晶と非結晶(アモルファス)の混合物から結晶化度の計算方法34)

XRD での結晶多形(或いは未知物質)の定量分析は、図 33 に示す結晶化度計算式で同様にして実施することが出来る。結晶多形の同定(定性分析)は、図 34 a) で示す様に、XRD スペクトルの比較により、ピークのパターン及びその強度等から既知物質(或いは結晶多形)かどうかの同定、或いはピークパターンから試料が混合物であるかどうかも判断できる24)。結晶多形の同定(定性分析)は、XRD スペクトルの比較によりピークパターン及びその強度等から既知物質(或いは結晶多形)かどうかの同定、或いはピークパターンから試料が混合物であるかどうかも判断できる24)。実際、図 34 b) に示すMebendazole の結晶多形の粉末 X 線回折(XRD)スペクトルであるが、結晶多形混合物(Form A + C)は結晶形 A と B の吸収ピーク及び強度パターンが合わさったスペクトルを与えている。Form A と Form Bは粉末 X 線回折(XRD)スペクトル上の2q = 6-17°付近に 2q 角が異なるそれぞれ1本のピークが与えるが、混合物はそれぞれの結晶多形のピークが合わさった2本のピークを与えている。この様に得られたスペクトルを比較することにより容易に同定・区別することが出来る。
また、図 34 c) はテオフィリン無水物中のテオフィリン水和物の含量を定量分析する方法を示している36)。例えば、ある結晶多形に混在する他の結晶形の定量は XRD スペクトル上で重ならない(影響しない)特異ピークを選び、混合している結晶形の結晶を一定量ずつ添加し得られるピーク強度から検量線を作成する。その検量線を用いて a : b 比から計算できる。疑似多形であるテオフィリン 1水和物 (18) とテオフィリン無水物の粉末 X 線回折のスペクトルであるが、疑似多形同士のピークが重ならない特異ピークを見つけ、添加量とピーク強度から検量線を作成し、試料に含まれる結晶多形の混合比(a : b 比)から混合量を計算することが出来る。
図 34. 粉末 X 線回折(XRD)法による定性試験(同定)と定量試験24)
a) 定性分析(結晶多形及び未知物質の同定)24)

b) Mebendzole の結晶多形の混合物の粉末 X 線回折スペクトル35)

35) Sara B. Honorato, et al., J.
Braz. Chem. Soc., 23(2), 220-227 (2012)
c) テオフィリン無水物中の微量疑似多形(テオフィリン 1 水和物)の定量分析(混合物含量の定量)36)

化合物 5 の結晶多形は、特許1) に従えば 3 種類(Type A、Type B 及び Type C)が確認されているが、図 35
に示す様に、それぞれの結晶多形の XRD スペクトルは特有形状のピークパターンを与え区別と同定を行うことが出来る。
図 35. 化合物
5 のXRD1)

下図 36 に示す様に、ある原薬の市販品1~4が原薬1形、市販 5~6 が原薬 2 形と帰属されている36)。しかし、それぞれ市販品は と で 様に、市販品 1~4 は原薬 I 形にないピーク、市販品 5~6 は原薬 II 形にないピークを有しており、それぞれは純粋に原薬 1 形及び 2 形ではなく他の結晶形の混じっていると考えられる(図 36)37)。
図 36. ある原薬のXRD スペクトルによる原薬の結晶多形の特定37)

37) MST 材料科学技術振興財団 分析事例C0473 2017/07/07より引用
3) 熱分析(DSC)スペクトルの読み方
抗てんかん剤として用いられるカルバマゼピン (12) には I 形晶、II 形晶及び III 形晶が知られているが、図38 は I 形晶のDSC結果である。175℃付近に吸熱ピークが現れた直後に発熱ピークが表れている。これは Ⅰ 形晶が融解の後に安定形である Ⅲ
形晶へ再結晶化(安定晶への転移)したためである。190℃付近の吸熱ピークは安定晶である Ⅲ 形結晶の融点(融解)である。その後、210℃ まで熱移動が観測されていないため、カルバマゼピンは 210℃ まで安定性が確保され分解しないことが読める(図 37)11)。
図
37. カルバマゼピン
(12) の DSC スペクトル11)

化合物 5
は最低 3 種類の結晶多形が存在している。それらの多形の融点(熱吸収ピーク)はDSC 測定から130.1、141.4 及び 149.0℃ にあり、TYPE A の結晶は130.1、141.4 及び 149.0℃ の 3 つの熱吸収ピーク(融点)を示し、TYPE C は141.4 及び 149.0℃ の 2 つの熱吸収ピークを示し、TYPE B は149.0℃ の単一ピークのみである。このことから、それぞれの結晶多形である TYPE A は不安定晶、TYPE C は準安定晶、TYPE B は安定晶であることがわかる。化合物 5 の不安定晶は、不安定晶(TYPE A)⇒準安定(TYPE C)⇒安定晶(TYPE B)へ熱転移していることがわかる。 化合物 3 の安定晶は融点での溶融後、大きな発熱が見られることから分解が起こっていると考えられる(図 38)1)。
図
38. 化合物 5 の
DSC スペクトル1)


化合物 6
は7) DSC による測定条件として 20℃/min の加熱速度で昇温すると多形 YN
はCで発熱し R へ、更に Y へ転移する(図 39)。DSC の昇温速度を 10℃/minとゆっくり昇温すると 20℃/minの昇温速度で観測できなかった多形 R、Y、OP及びONの融点が確認出来ている。更に、結晶形 R は昇温速度を5℃/min にすると106℃ 付近の融点を示し、106.5℃ 付近で発熱ピーク(結晶化熱)が見られ熱媒介転移を起こし結晶形 Y へ結晶化し、110℃付近で結晶形 Y の融点、更に、111℃ 付近の発熱ピーク(結晶化熱)により結晶形 OP へ、しかし、OP から結晶形 ON への結晶化熱は認められないが、それぞれの結晶多形の融点が観測されている1)。この様に、DSC 測定時に昇温速度をコントロールすることにより、結晶多形の熱媒介転移(結晶化熱)を観察することが出来る。
図 39. 5-Methyl-2-[(2-nitrophenyl)amino]-3-thiophencarbonitrile (6) のDSC 測定7)


図 40
は、結晶多形ではないが疑似多形である化合物 17 の DTA と TG を組み合わせた熱分析結果である38)。このスペクトルは何を意味しているか?DTA と TG を組み合わせた熱分析のスペクトルは熱による化合物の重量変化と結晶の状態変化を同時に示すことが出来る。
図 40. 疑似多形である化合物
17 のDTA
と TG を組み合わせた熱分析スペクトル


化合物 17のDTA スペクトルは 98.6℃ に第一熱吸収ピーク、157.1℃ に第二熱吸収ピークと 175℃ 付近の発熱ピーク、及び 230.2℃ に第三熱吸収ピークを示している。TGスペクトルは 85~105℃ 付近で 3.8% の重量変化(重量減少)と 271.9℃ 以降に連続的に重量減少が示している(図 41 左)。
本スペクトルから、TGの 85℃付から観測される重量減少は本化合物の 1 水和物量(重量の 3.8%)に匹敵している。DTA スペクトルの第一熱吸収ピーク(98.6℃)は脱水(蒸発)による熱吸収と考えられる。このことから、本化合物は 1 水和物であると考えられ、更に昇温していくと 157.1℃ 付近で結晶の溶融による熱吸収とそれに続く発熱ピーク及び 230.3℃ 付近の熱吸収が観測されている。この一連の本化合物の熱吸収と発熱は、98.6℃ で結晶中のパッキング様式が変化せず結晶中の分子から 1 水和物量の水分が抜けただけの不安定メタンスルホン酸塩の結晶、第二熱吸ピーク 157.1℃ と175℃ 付近の発熱ピークは不安定メタンスルホン酸塩の融点と結晶中の分子のパッキング様式が変化して安定メタンスルホン酸へ転移した結晶化熱と考えられる。また、第三熱吸収ピーク 230.2℃は安定メタンスルホン酸塩の融点と考えられる。実際、メタンスルホン酸塩の単結晶を作成し単結晶 X 線構造解析と融点を測定した結果、230℃付近に融点を持ち、化合物 17 (1 水和物)と異なる単結晶構造を持っていた(図 41 右)。不安定なメタンスルホン酸塩の結晶は作製することが出来なかったが、安定なメタンスルホン酸塩と融点が異なることから、存在していたと考えられた(図 41 下)。
図
41. 化合物
17(水和物・メタンスルホン酸塩)の
DTA-TG スペクトルから分かること38)

安定メタンスルホン酸塩 (18) のDTA-TG スペクトル

4) 個体
13C-NMR スペクトルの見方
固体13C-NMRによる結晶多形の同定はスペクトルのピークのケミカルシフトと強度及び緩和時間の違いから行うことが出来る。
例えば、チアミン塩酸塩 (20) は Ⅰ 形晶と Ⅱ
形晶の結晶多形が存在することが知られている39)。チアミン塩酸塩の結晶多形は固体13C-NMRスペクトル上で幾つかのピークのケミカルシフトが明らかに異なっており、それぞれの結晶形を区別、同定することが出来る(図 42)。
図 42. チアミン塩酸塩(20)の結晶多形の 13C-NMR スペクトル39)

インドメタシン (2) は安定結晶である γ 形晶のみ単結晶構造解析が行われているが、α 形晶に関する報告はない。インドメタシン (2) を固体 13C-NMR で測定すると、図 42 に示す様に39)、γ 形晶は各炭素に1本ずつのピークが観測されるのに対して、α 形晶では各炭素に複数本のピークが観測されている。インドメタシン (2) の OCH3 と CH3
のピークを拡大すると OCH3 は 4本のピークに、インドリン環上のCH3 は3本にピークに分かれている(図 43)。このことから、インドメタシン(2)の結晶多形は固体 13C-NMR のスペクトルを比較することにより、区別・同定することができる。また、g 形晶は 13C-NMR スペクトル上で分子内のそれぞれの炭素が単一のピークを与えるため単一の分子コンフォメーションによりパッキングされていると思われる。しかしながら、a 形晶中には、 13C-NMR のスペクトルから 3 種類以上の分子コンフォメーションを取った分子がパッキングされていると考えられる39)。このことが、a 形晶の単結晶構造解析がなされていないものと思われる。
図 43. インドメタシン (2) の結晶多形の
13C-NMRスペクトル39)

図 44
に示す様に、インドメタシン (2) の非結晶(アモルファス)は分子の配列(パッキング)がランダムであるため、固体 13C-NMR スペクトル上でブロードピークを与える40)。
図 44. インドメタシン
(2) の非結晶(アモルファス)のNMR スペクトル40)

テルフェナジン (21)は form Iと form II の結晶多形を有しているが、それらの結晶多形の固体 13C-NMRスペクトルはほぼ同じケミカルシフトとピーク強度を与える(図 45)。スペクトル上のケミカルシフト 31~32 ppm 付近(t-Bu-CH3)を拡大すると分離している39)。このことから、化合物21の 2つの 13C-NMR スペクトルは結晶多形を同定することが可能であることを示している。また、結晶多形である Form I と II は NMRスペクトルから結晶中分子のコンフォメーション及びパッキング状態が非常に近似していることを示していると考えられる。
図
45. テルフェナジン(21)の固体 13C-NMR スペクトル39)

この様に、得られた各種分析機器のスペクトルは、スペクトルを重ね合わせる、拡大させる等により注意深くスペクトルを読むことによって結晶多形中の分子の状態を予測することが可能となる。得られたデータは慎重に、大切に取り扱うことを進める。
終わりに
医薬品原薬の結晶多形は医薬品にとって非常に重要な品質であるため、PMDA(医薬品医療機器総合機構)は医薬品の承認(或いは、MF)申請時に結晶多形について記載を求めており、その原薬の結晶を見分ける方法(分析)を知ることはプロセス研究者にとって大切であると考えている。
結晶中の分子コンフォメーション(分子立体構造)と分子パッキング(分子配列)の違いは、溶媒分子と化合物分子との相互作用により分子コンフォメーションが、化合物同士の分子間相互作用(水素結合、π-π及び CH-π 結合等)による分子パッキングの違いが結晶の物性の差として現れる。このことから、結晶分析出来る機器のスペクトルの違いとして現れる。既にお話しした様に、結晶多形は固体状態で分析できる機器である IR (Raman) spectrum、粉末 X 線回折、DSC(示差走査熱量測定:Differential scanning calorimetry)並びに個体 NMR等で実施することが出来る。これらの分析機器の中で、IR (及びRaman) 及び XRD(粉末 X 線回折) は結晶多形の同定(定性分析)及び定量分析が出来る。 IR はポピュラーな分析機器であり結晶多形の同定と定量(混合比等)に用いられる。更に、固体 13C-NMR及びXRDは結晶と非結晶(アモルファス)を見分けることが出来る。しかしながら、基準物質として純品の各結晶多形の結晶がない場合は、結晶形の同定と定量は困難となるが、熱分析である DSC(或いは、DTA) と組み合わせると、熱の吸収と発熱を測定でき結晶の融点、結晶加熱化熱、相転移、分解熱等が測定できるため、安定晶、準安定晶或いは不安定晶等を見分けることが出来る。DTA-TG(熱重量分析 (Thermogravimetric Analysis))があれば結晶の重量変化も同時に測定できるため、疑似多形である結晶に含まれる水(溶媒)が水和物(溶媒和物)か付着水(付着溶媒)かの判別、並びに脱水後の結晶転移、分解による重量変化等が測定出来る。
筆者が大学の研究室で有機合成化学を学び始めたころ、先輩から「研究は分析に始まり分析に終わる」と言われた。その時は、この言葉がピンと来なかった。しかしながら、製薬メーカーで創薬研究を始める化合物合成と分析(HPLC, IR, NMR, Mass, UV)、HPLC による分取、それらを用いた構造決定(IR, NMR, Mass, UV, XRD)、並びにプロセス開発での反応中、中間体、不純物及び原薬等の分析(HPLC, IR, NMR, Mass, UV, DSC, Powder XRD)を実施していると「研究は分析に始まり分析に終わる」の言葉はもっともであると納得した。プロセス研究者は、結晶多形の分析が医薬品の原薬にとって非常に重要なことを認識し、基本的な分析法・各種分析機器から得られる各種スペクトルを解析する力を持つことが非常に大切である。
本内容がプロセス開発を行っている方々の参考になれば幸いである。結晶多形の分析方法並びにスペクトルの解析について詳細に知りたい方は、各分析機器の分析専門家に聞くか、web 上での検索、専門書等により知識を得て頂きたい。
コメント
コメントを投稿