
表 1 開発ステージでのプロセス開発
検討場所
|
検討段階
|
数量(kg)
|
検討内容
|
実験室
|
小スケール実験
|
~0.10
|
反応条件を含む工程操作の最適化
・合成ルート、反応条件、合成法の開発
・出発物質(原料)、副原料、試薬、触媒及び溶媒等の種類と量比の
最適化
・反応温度を含む工程の操作温度の最適化と許容範囲の設定
・合成法の最適化,
・規格(品質)及び試験方法の開発
・原料、試薬、副原料、溶媒等及び中間体・原薬の物性確認
・精製方法(抽出、晶析、カラム、蒸留等)の開発
・原料、試薬、副原料、溶媒等の品質規格の設定
・中間体・原薬の品質目標(品質規格)の設定
・品質リスクマネージメントに従いデータ取り
|
サンプル合成
|
~10
|
・小スケール実験で最適化した合成(製造)法の妥当性の評価
・原料、試薬、副原料、溶媒等の品質規格の妥当性の確認
・及び中間体・原薬の目標品質(品質規格)の妥当性の確認
・原薬の品質特性の確認
・ジェネリック会社のサンプル評価
|
|
品質
リスクマネージメント
|
品質リスクマネージメントとして、
・品質リスクアセスメントの実施、パラメータのデータ取り計画と実行
・合成ルート及び合成(製造)操作パラメータの評価
・品質リスクマネージメントに従いデータ取りと重要工程、クリティカル
パラメータの把握
・得られたデータ解析から品質に影響を与えないパラメータの許容範囲、
安全率を掛けた操作範囲並びに実際に操作する目標値の選定
・スケールアップシミュレーションとスケールダウンシミュレーション
・原料・試薬・副原料・溶媒等及び中間体の品質規格の妥当性評価
・原薬・中間体の規格及び試験方法の妥当性の評価
|
||
パイロット
プラント |
パイロット製造
スケールアップ実証 |
~100
|
・実験室で最適化した製造法(標準製造法)のスケールアップの妥当性
の評価
・パイロットで使用した設備・機器(材質・機能・性能等)の妥当性の評価
・仕込み、反応、撹拌、濃縮、ろ過及び乾燥等の工程操作の状態確認
・工程内の各操作に要した時間、目標温度の維持状態等の確認
・得られた原薬の品質の確認
・原薬の純度、不純物プロファイル、不純物量、性状等の品質規格項目への適合性の確認
・更なるスケールアップへの問題点の抽出
|
データ解析
|
・パイロット製造での工程単位操作(仕込み・反応・後処理・抽出・濃縮・
ろ過・乾燥の温度と時間と安定性)の再現性確認
・反応状態、撹拌効率等の相似性の確認
・加熱・冷却温度の制御方法と時間の再現性の確認
・不純物の生成メカニズム、不純物の制御方法と低減方法(反応条件と
精製法)の解明及び開発
|
||
製造
プラント
|
GMP 製造
|
~500
|
GMPを遵守した製造法、臨床試験用の原薬の供給
・GMP、当局の規制に従い製造法の確立
・工程操作パラメータの操作範囲並びに目標値の妥当性(維持の可能性及び品質への影響)の確認
・スケールアップに耐えられる標準製造法の確立
・中間体及び原薬の取扱法の確立
・臨床試験への治験薬(原薬)の供給
・標準製造法の堅牢性の向上
|
品質リスクアセスメント
製造法・設備 治験薬製造 |
Process
Validation (PV) 実施前に、
・今まで得た全てのデータ及び経験からスケールアップによる品質、
危険性、安全性等についてのリスク評価を実施
・クリティカルパラメータの選定とPVでの検証項目を設定。
・単位操作の不具合の発生確率と詳細な検討の要否
・使用予定設備のインパクトアセスメントと適格性評価検証確認
(一般的には,既存設備は適格性評価が終了していることから、Process Qualification (PQ) での検証となる)
|
||
商業生産
プラント
|
プロセス設計:
Validation Master Plan
(VMP) と
Process Qualification (PQ)
Process Validation(PV)
|
~1,000
|
Validation
Master Plan (VMP)
・Validation Master Plan (VMP)の作成
・Process
Qualification (PQ) 計画書作成と実施並びに報告書
・SOPと製造設備の構造・機能・能力の検証
・Process Validation(PV):
・検証範囲の特定と標準操作法の連続 3バッチでの妥当性の確認
(PV製造)
・標準製造(商業製造)法の確立
|
商業製造
|
~1,000
|
・商業生産運転を通じて商業製造法の検証(アニュアルレポート)
・得られた原薬が恒常的に品質規格に適合するかの確認
・原薬の品質及び収量の傾向分析
・製造法の堅牢性の確認
|
|
コスタダウンの検討
|
~1,000
|
・アニュアルレポートによる品質・収量向上に向けた商業製造法の最適
化(操作範囲の厳格化、目標値の運転)
・商業製造法の最適化のトレースできる設備・機器への変更
・中間体へ出発物質変更
・更なるスケールアップ
・新規製造法の開発によるコストダウンへ
|
投入はスケールアップに伴い取り扱う原料等が膨大となり、危険性(発熱・静電気爆発)・有害性(吸入・接触等による吸収)
等が増大する。

この時の教訓として、製造前に使用する主原料、副原料、試薬、触媒、溶媒等に至るまでSDS等を、また、受託化合物であれば委託先からSDSを取得して危険性・有害性・毒性等の調査を実施すべきと考えている。調査で得られた危険性・有害性・毒性に対する事故時の処置法並びに有害事象が発生した場合の治療法等を製造前打合せで共有していた。大量の危険性・有害性・毒性化合物を取り扱う時は、投入作業時の設備(封じ込め、拡散防止)と作業者の装備(手袋、メガネ、マスク、作業服の種類等)の選択、更に、作業中に何かあれば即座に報告することを徹底させ安全な対策が確認されるまで製造作業が出来ないことなどの取り決めを持っておくべきです。
実験室での反応時の撹拌数(回転数(rpm))を確認していると思うが、実機での撹拌数を正確に反映させていないのが現実であると思っています。今までの経験から製造現場で撹拌数を設定してほぼ実験室を再現出来ていたからだと思う。それは、たまたま反応が均一系、加熱、二相反応でも実機の最大撹拌で事なきを得ていただけだと思っています。



抽出では、液―液間での分配が重要となるため撹拌による滴-滴のサイズを小さくして比表面積を大きくし滴滴の接触回数を増やすことで抽出効率を上げることが出来る。濃縮では濃縮槽の溶液上部表面から溶媒蒸気が上がるため濃縮液の表面を常に新しくすることにより濃縮速度を上げることが出来る。


製造操作法の条件・数値の示強性変数(変更してはいけない数値:原料と副原料・試薬
例、ある粗原薬の造塩(マレイン酸)による精製工程で、晶析率が上がらない事態が発生


操作
|
実験室
|
製造現場
|
||
1.原料・副原
料・試薬等の確認
|
実験室での原料・試薬・副原料・触媒等

・原料(出発物質)、副原料、試薬及び触媒等はユーステストで得られた中間体及び原薬等が品質規格に適合する品質の原料等を選択する。
・出発物質が委託合成品の場合、出発物質の妥当性(出発物質の一般原則)を満たす必要がある。
・選択した原料(出発物質を含む)・副原料・試薬等の品質規格及びその試験方法を設定する。
・試験方法は原料・中間体並びに含まれる全ての不純物が分析できる方法でなければならない。
|
製造現場での原料・副原料・試薬等

・原料・副原料・試薬等は汎用性のある工業薬品をなるべき選択する。医薬品専用原料(化合物)の場合は、出発物質等の妥当性を確認する必要がある。
・製品標準書に記載されていないメーカーの原料(出発物質を含む)・副原料・試薬・溶媒等を使用してはいけない。
・使用する全ての化合物の危険性、安全性並びに環境リスクアセスメントを実施し、危険性に合わせて仕込み方法、保護具選択等を考える。
|
||
2.溶媒
|
実験室での溶媒
 ・溶媒には反応、抽出、共沸、晶析及び洗浄溶媒があり、それの選定と使用量の決定を行う必要がある。
・溶媒の種類が決まれば、医薬品原薬製造グレード(ドラム・タンクローリー)の溶媒を用いて最適反応条件、中間体・原薬品質の確認のためユーステストを行う。
・溶媒の純度・脱水率はタンクローリー>ドラム缶>試薬瓶の順に悪くなる。
・反応溶媒種は工程反応の反応速度、反応濃度(~2 mol/L)及び生成物の品質(純度・変換率・不純物プロファイル等)の検討により選択する。
・抽出溶媒は生成物の溶解度(~5
w/w)・反応(分
解)性・危険性・精製度合いから設定する。
・晶析溶媒は溶媒量(~8 w/w)・精製度(純度・不
純物プロファイル・回収率等)の最適化検討と反応槽等の容積率、晶析濃度を設定する。
|
製造現場での溶媒

・使用溶媒は大量入手が可能で、安定供給出来、医薬品原薬の使用に耐える品質が必要である。
・原料(出発物質)等との溶媒量比(濃度)は変更
してはいけない。
・反応濃度は反応速度と仕込み効率(経済性)に影響するため、1 mol/Lを基本として混合溶液の撹拌状態(粘度)により濃度を調製する。
・一般的に、新品のドラム缶入り溶媒はグリニャー
ル反応に使用可能である。
・プロセス開発で設定した操作条件に合わせ仕込み原料量に合わせ溶媒量・溶媒比・仕込み率を正確に秤量し仕込む。
・選択された反応・抽出・晶析等の溶媒の危険性・
有害性等を確認し、注意する必要がある。
・安全環境リスクアセスメントからの対策と徹底。
|
||
3.反応機・
晶析機
|
丸底フラスコ或いは幾何学的相似形反応容器
実験室 ~2 L反応装置

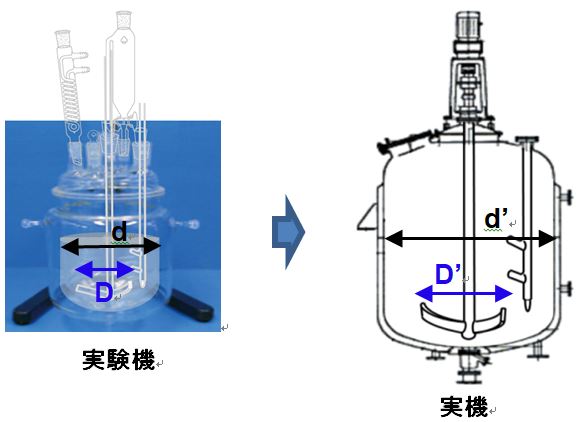
・実験室では、一般的にガラス製丸底フラスコと半月板撹拌翼付き実験機を用いてデータを取ることが多いが、現場の実機反応槽と形状が異なる。
・実験室の実験機と製造現場の実機反応槽・晶析槽の撹拌・反応・混合状態を相似させるためには、製造で使用予定の実機をスケールダウンした幾何学的相似形ガラス反応槽を用いることを薦める。
・幾何学的相似形反応槽を用いても、得られた全てのデータをそのままスケールアップしようとしても困難な場合(撹拌状態・伝熱効果)がある。
|
実機反応槽・晶析槽の選択
製造現場 ~10,000 L反応機

・実機反応槽等は反応条件・工程操作に最適な性能・能力及び材質から選択する。
・プロセス開発時と同等以上の中間体・原薬の品質(純度・不純物プロファイル・不純物量・結晶サイズ等)及び収率を再現するためには、操作条件の示量的数値を使用予定実機のサイズに合わせ変更する必要がある。
・特に、撹拌数及び伝熱効果を反応槽のサイズに合わせて計算する必要がある。但し、均一系では極端な撹拌数の違いがない限り大きな影響を与えることはない。
|
||
4.仕込
|
原料:~200 g、溶媒:~5 L
|
原料:~1,000 Kg、溶媒:~10,000 L
|
||
原料・溶媒等の投入方法
|
実験室での仕込み方法
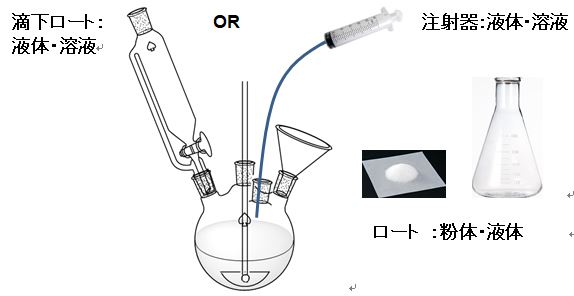
・粉体、液体或いは溶媒に希釈した溶液か、不活性ガスケア必要か、仕込み方法は直接投入か、減圧吸引、圧送、ポンプ移送或いはその他かの選択。
・試薬・副原料等の投入(仕込み)時の発熱と仕込み時間が反応速度及び目的物の品質に与える影響を確認する。
・中間体、原薬、原料(出発物質を含む)、副原料並びに試薬等の反応条件下で熱安定性を確認する。
・中間体・原薬・原料(出発物質を含む)・副原料・試薬等の仕込み時間による熱安定性の確認。
・可燃溶媒・粉体等による静電気の危険性評価と対策とSDS(特に、変異毒性等)等により、原料、副原料及び試薬等の危険性・安全性を確認する。
・安全及び環境リスクアセスメントを実施し、作業者・環境への配慮も忘れてはならない。
・医薬品中間体・原薬等の製造は、製品標準書に記載された標準製造法に従い工程を遂行させる必要がある。
|
製造現場での仕込方法

・プロセス開発で設定した仕込み方法・仕込み時間を実現させる設備(現場設備の性能・能力の把握)を選択し、熱安定性が確保された時 間内に中間体、原薬、原料(出発物質を含む)、副原料並びに試薬 等を仕込む。
・但し、製造操作法にない状況、突発的な事故等が生じた場合は中止 する勇気を持つ。
・仕込み作業は、全ての原材料が大量となるため、作業者への暴露と 静電気爆発等が最も懸念される。このことから、危険性、安全性、保護具の選択並びに環境等に対する教育と対策を徹底する。
・可燃溶媒・粉体等による静電気の危険対策としてアースの設置等を実施する。
|
||
5.反応
|
加熱・還流・冷温・極低温・高温・水素添加・加圧・二相反応等
|
加熱・還流・冷温・極低温・高温・水素添加・加圧・二相反応等
|
||
反応条件
|
一般的な反応装置、撹拌翼と伝熱面積
・実験室の反応装置は丸底フラスコ-半月板撹拌翼が一般的に用いられるが、製造実機と構造-形状が異なる。実機の幾何学的相似形反応装置は構造-形状が同じであり、反応条件のスケールダウンシミュレーションで実験室と製造現場を繋ぐ検証に有用である。
・反応装置、撹拌翼及びバッフルの構造-形状により伝熱面積と撹拌状態が違ってくる。

反応条件の検討
・反応条件検討では、原料と副原料・試薬・塩基・酸・触媒等の量と比率、液性、溶媒種類、反応温度、滴下速度及び撹拌状態等操作範囲/操作条件を最適化させ反応速度、目的物の変換率、品質、不純物量及び不純物プロファイル等を検討する。
・反応条件の検討では一次検討としてパラレル反応装置等を用いて検討すると、効率よく実施出来る
・二次検討として丸底フラスコ或いは現場実機の幾何学的相似形反応槽(推奨)を用いて一次検討で得た反応条件を最適化する。
・次に、二次検討により反応条件が最適化できれば予定スケールに合わせた滴下速度、加熱・冷却能力、撹拌数等を実機の性能・能力を基に予測計算する(スケールアップシミュレーション)。
・反応条件(温度・圧力)と反応終了時間並びに操作予測時間を加味した熱安定性のデータ取りと熱安定性の結果から反応条件の操作許容範囲・操作範囲を設定し、実際の操作条件を決定する。
・このようにして得られる中間体・原薬が恒常的に品質規格に適合させるために、スケールアップシミュレーション及び幾何学的相似形反応装置を用いたスケールダウンシミュレーションにより反応条件を最終調整する。
・反応の確認には、TLC法とHPLC法がある。反応条件の一次(予備)検討では、パラレル合成機を用いて反応条件をTLCにより予備検討する。二次検討では、反応条件である反応温度、反応溶媒の種類と量、圧力、及び原料と試薬等の量比等を最適化するためにHPLC等を用いてコストと品質に関わる変換率、純度及び不純物量等を定量的に取り扱う。
・注意、TLC法は反応混合物中の全ての化合物を分析できるが定量的に扱えない。HPLC法は検出ピークを定量的に取り扱えるが反応混合物中の全ての化合物を分析できるとは限らない。従って、HPLCの試験方法は全ての反応混合物が検出できる条件でなければならない。
実験室での予備検討の反応チェック(TLC法)
HPLC法による反応のチェック(例)
・反応チェックはTLCで反応速度の当たりを付け、反応状態の推移は、HPLCの分析時間を考慮し、サンプリングを1時間毎或いは1, 2, 4,
8, 16, 24, 36----hr毎に行い分析する。
実験室でのサンプリング例
 
反応終了時間が5時間程度と分かっている場合は、反応後4時間目から1時間毎に、また7~8時間までサンプリングして反応混合物の推移を確認する。
・反応終了確認後もHPLCで分析し、目的物、不純物等の変化を確認する。
・反応でスケールアップ時に問題となるのは、特に、伝熱効果、撹拌 効率及び操作時間である。
・スケールに合わせた製造現場での操作時間を予測し、時間と温度による原料、中間体、生成物等の熱安定性を担保するためのデータ取りを実施する。
・反応、原料・試薬・溶媒等の危険性(静電気爆発性、熱の蓄積性、反応の暴走性)、安全性(作業者の健康へ影響)及び環境への負荷を調査する。
|
一般的な反応槽、撹拌翼と伝熱面積
・実機には、Glass Lining (GL)とStainless Steel (SUS) 、HASTELLOY*、樹脂製等がありバッフルと撹拌翼の構造-形状が異なる。これらの選択は反応液の液性(酸性或いは塩基性)或いは高温-低温反応条件等で行う。
・SUSとGL製反応槽の構造-形状の差が撹拌及び伝熱状態の違いとして現れる。

・反応条件により専用(極低温、極高温、酸性用、塩基用反応槽及びユーティリティーの性能・能力)の製造実機を選択する。
標準製造法(操作法)のトレース
・プロセス開発で設定した工程の反応条件は製造現場(GMP製造)で変更してはならない。
・原料・副原料・試薬・溶媒等の量はスケールに合わせて変量するが、それらの量比は変更してはいけない(示強性と示量性変数に注意)。
・プロセス開発で最適化した反応条件である温度、圧力、濃度、量比、時間及び撹拌状態(示強性変数)は変更してはいけない。それ以外の条件(示量性変数)はスケールに合わせて変更する。特に操作時間はスケールに合わせ増大するため、プロセス開発時に安定性の担保が重要となる。

・但し、パイロット製造(non-GMP)では製造操作状況を確認しながら進める。プロセス開発で設定した条件が製造現場で再現できない場合は、現場実験として操作条件を変更することがある。その時は実験室で原因を究明しプロセス開発をやり直す。
製造現場での反応終了確認
・反応完了試験は、プロセス開発で設定されたHPLC試験方法(分析)に従って実施する。
・この時、反応速度、反応目的物の変換率、品質と不純物プロファイルがプロセス開発と同等かそれ以上の品質であることを確認する。
HPLC法による反応のチェック
プロセス開発時に得られた反応終了時間の1時間前からサンプリングしHPLC分析により反応進行状況と反応終了を確認する。反応終了が確認されたら反応停止操作に移る。
反応チェック(例)
反応終了が5時間程度の場合

プロセス開発で設定した反応終了確認試験条件(HPLC)及び反応完了規格に従い反応の終了を判定する。
製造現場で品質に影響を与える操作
・プロセス開発で得られた最適な操作条件、操作時間、撹拌状態及び伝熱状態等をスケールアップ後の反応槽で忠実に再現出来れば、プロセス開発で得られた同等以上の品質・収率で中間体・原薬を得ることが出来る。しかし、実験機内の状態を忠実に再現することは難しい。だから、化学工学の学問がある。
・例えば、50 Lから500 L反応槽への10倍のスケールアップ後の伝熱面積は5.5倍程度しか増えない。実験機と実機では加熱-冷却(伝熱効率)に時間を要する。
・次に、撹拌状態の違いは反応今後応物が不均一となり反応速度、品質-不純物プロファイル等に影響を与える。撹拌数のスケールアップはPv(単位体積当たりの撹拌動力)一定でスケールアップすると実機反応槽でほぼ同等に再現できる。
・反応及びその他の操作は、熱安定性が担保されている時間内に操作・作業が終了させる必要がある。そのためには、製造を有する設備の選択と操作・作業の見直しを実施する。
一般的な実機での操作時間(一例)
時間は各社製造実機の性能・能力により異なる。

・反応に関する危険性、安全性及び環境への負荷については、現場作業者への教育を徹底する。
|
||
冷却・加熱
|
実験室での加熱・冷却に用いる装置
・加熱及び冷却は、温水、オイル或いはマントルヒーターによる加熱、冷水、氷-食塩或いはドライアイス-溶媒(~-75℃)等により行われる。
実験機での加熱-冷却
加熱及び冷却は水浴、マントルヒーターにより行われる。
加熱

冷却
 実験反応槽の伝熱面
・伝熱面積/槽容量比が大きいため加熱・冷却は速い(例えば、50L槽の伝熱面積:0.51m2*(L当たりの伝熱面積:0.0102 m2/L)

・特に、冷却時の伝熱能力・必要な冷媒温度の確認とデータ取りを行い、スケールアップ後の伝熱能力と冷媒温度を予測計算する。
・反応温度は10℃刻み(必要に応じて~5℃など)で最適化、反応温度は幅で設定し、仕込み時、反応中等の温度推移を確認する。
・収率と不純物プロファイルを確認する。
・反応危険性の評価(設定温度に+20℃の危険性)を行う。
・発熱反応では、総発熱量を確認する。
・予備計算としては、一般的なSUS, GL反応槽の総括伝熱係数を用いることが出来る。
伝熱計算関係
反応槽の伝熱面積(八光産業の技術データ):http://www.hakko-sangyo.co.jp/wp-content/uploads/2013/10/ab4b9ed0c028ef45ad95d6ac33f1b132.pdf
総括伝熱係数:
ジャケット&攪拌機付きタンクの伝熱計算Ver1.1(ライセンスキー必要):
|
・実機反応槽では、加熱は温水、高圧蒸気(~135℃)、低圧蒸気或いは高温オイル(~250℃)による加熱と冷水、冷媒(エチレングリコール、液体窒素或いは液体窒素によるアルコールの冷却等;~-90℃)により行う。
実機反応槽での加熱-冷却
実機の加熱冷却はジャケットに熱媒或いは冷媒を通すことにより行われる。
反応槽ブライン切替型加熱・冷却方式*

実機の伝熱面積と加熱・冷却計算シート
・伝熱(ジャケット)面積/槽容量比が実験室より小さくなるため、加熱・冷却に時間が掛かる。
  ・例えば、5,000L槽では12.85m2*(L当たりの伝熱面積: 0.00257m2/L、50L槽と同等の伝熱(ジャケット)面積/槽容量比を確 保するためには51.0m2の伝熱面積が必要となる)
・プロセス開発データから予測した必要伝熱能力と冷媒温度が可能か を実機製造設備・機器の能力を机上で検証し実機を選定する。実際 に、選定した製造設備を用いてスケールアップ製造を行い検証す る。
加熱・冷却時の特定温度への到達時間計算式:
但し、スケールによる冷媒温度の⊿tに差が無い時の所望温度への簡易到達時間計算シートである。
間計算から生成物等の熱安定性及び冷媒温度等を確認するために、 伝熱計算式を組み込んだシートを作成してもらった。
・一般的な総括伝熱係数

㈱神鋼環境ソリューション(http://www.kobelco-eco.co.jp/product/process/docs/glass_tech_1-4.pdf)より引用
|
||
試薬・副原料等の投入方法
|
製造量: ~500 g
適下ロート・定量ポンプ等を使用
・実験室では、溶液の試薬或いは副原料等の滴下は滴下ロートで行うのが一般的である。

温度計は安全性とデジタルで表示できる熱電対、測温抵抗体に切り替わっているが、ここでは便宜的にガラス製(水銀)で示している。
・滴下ロートを用いることが多いが、低温で試薬等を滴下する場合はジャケット付き滴下ロート或いは無水反応の場合は試薬を冷却しカニュラ―で投入、定量的に投入する場合は、シリンジ
・スケールにより投入方法を変更する必要が出てくるが、投入方法・投入時間と原料・生成物等の安定性が問題となる。また、小スケールでは静電気の問題はないが、大スケールになると問題になる。
スケールアップ後のスケールに合わせて投入方法を考える。
|
製造量: ~1,000 Kg(100倍~1000倍)
ダイヤフラムポンプ・滴下槽等を使用
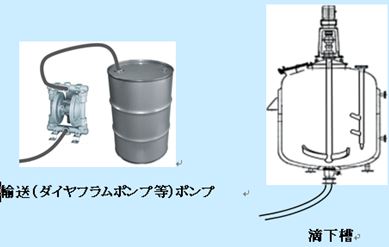
・製造現場では、溶液の試薬或いは副原料等の滴下は滴下槽、ダイヤフラムポンプより滴下される
 ・実機反応槽へは、溶媒はタンクからライン或いはドラムから、原 料・試薬等は固体の場合フレキシブルコンテナ―で、液体はドラム 或いは滴下槽から行う。
・低温等で試薬等を滴下する場合、ジャケット・撹拌機(翼)付きの 滴下槽が必要となる。
・静電気は、物質が移動すると発生する。特に溶媒存在下に粉体を投 入すると静電気が発生し溶媒に引火する可能性があるので、対策を 考える。
|
||
投入時間
|
・滴下は、滴下速度と発熱量を見ながら反応温度に合わせて行うが、少量では冷却効率が高く短時間で終了する。
・スケールに合わせた滴下時間の予測と目的物の品質、不純物プロファイル及び変換率等に影響を与えない滴下時間の熱安定性を確保する。
・スケールアップ時の滴下予測時間に1.5倍程度の熱安定性(目的物が規格適合出来る)を担保する。
・例えば、発熱が無い時、滴下速度:0.05L/minなら滴下容量0.3Lでは6 min 必要となる。
|
・製造現場での滴下時間は、製造スケールに関わる滴下する容量(製造量)、冷却効率(伝熱面積及び冷却剤量)から計算できるが、伝熱面積が小さいため実験室より増大する。
・プロセス開発で担保された滴下時間を満たすために、設備の選択と冷媒温度等を設定する。
・例えば、滴下容量が300Lの場合、滴下速度:10L/minなら30min必要となる(実験室より5倍の 時間を要する)。
|
||
発熱
|
・反応熱は原料・副原料・試薬・溶媒等の仕込み量が少ないため小さい。
・もし、発熱があっても大量の寒剤で冷却できコントロールしやすい。
・もし、コントロールが出来ない場合は、反応自体或いは条件を再考する必要がある。
・反応時発熱が激しい場合は、試薬或いは副原料を滴下する等により反応をコントロールすることを考える。
・原料・副原料・試薬・生成物の熱安定性と発熱量の確認及び反応が暴走しないかのデータ取りを行う。
・DSC、RC 1、ARC等で発熱反応の総発熱量・暴走温度等を危険性評価する。
|
・製造実機では、反応熱量は原料・試薬等の容量が増大するため莫大となる。
・反応熱がある場合、急速に寒剤を大量に投入できないため、実機で発熱をコントロール出来るかを発熱量からユーティリティー(冷媒温度、冷却機、等)の能力を検証する必要がある。
・反応が暴走する可能性がある場合、製造を中止するか、発熱の制御方法或いは反応条件(方法)を変更することを考える必要がある。
|
||
撹拌
|
撹拌翼形状:回転子・撹拌翼(半月板・幾何学的相似形)等
・実験機の撹拌モーター回転数:~1000rpm
・撹拌数は反応速度、発熱状況、目的物の品質、変換率、不純物プロファイルから設定する。
・撹拌は混合液を均一にする、反応温度を均一にする、反応熱を系外へ均一に排出させる、反応を均一に進行させる等の役割を担っている。
・一般的に均一系反応では撹拌による反応差は殆ど起こらないが、二相系反応(固-液、液-液)では撹拌状態(効率)が反応に大きな影響を与える。
・撹拌数最適化は、実験装置で実機最大撹拌数から計算した回転数以下で反応が進行し、最適撹拌得られるかを検証する。
・大小の反応槽で撹拌状態は「単位体積当たりの撹拌動力一定」でスケールアップすることにより同等性が確保出来ると言われている。

・撹拌数は目的物の純度・不純物プロファイル・反応速度等に最適な値とスケールアップ撹拌モーターの能力から設定する。
|
撹拌翼形状:三枚後退翼・バトル・アンカー・最近の形状撹拌翼(ツインスター®・MOLE PAW® )等
・一般的の実機の撹拌モーター回転数:~150rpm
・大小の反応槽で撹拌状態を相似させるためには、スケールアップ後の反応槽の翼径D(槽径:d)が長くなるため、回転数Nは少ない回転数でよい。
・大小の反応槽での反応速度を相似させるためには、撹拌状態を同じにする必要がある。予定実機反応槽の撹拌翼径(或いは、槽径)に合わせた実機の撹拌数は N’ =N X D2/3/
D’2/3 で計算と計算値が実機撹拌機の回転数範囲内かを確認する。
・大小の反応槽の撹拌状態を同一にするために、単位体積(容量)当たり撹拌動力(Pv)一定でスケールアップする。
Pv = N X D2/3 一定(=
N’ X D’2/3)
N:撹拌数、D:翼径(若しくは、槽径)
で撹拌のスケールアップするため、実機の撹拌数は少ない(実験機の撹拌数は多い)。
・実機の撹拌数がプロセス開発中に得た撹拌数の能力外の場合は、必要撹拌数を満たす別の反応槽へ変更か、撹拌機の改造により性能アップさせる。
|
||
反応時間
|
反応時間は基本的に設定せず、反応終了確認は工程内規格に従い実施する。
・反応時間を設定すると、反応速度は反応温度、濃度及び撹拌状態等により影響されるため、反応時間が延長する等により逸脱する場合がある。
・反応完了付近では、試験を1時間毎に行い不純物の生成状況を確認にすると共に、反応終了確認後も反応状態のまま延長し、反応終了確認試験で生成物の品質(純度・不純物量・不純物プロファイル等)及び収率の確認し、反応条件での安定性等の担保を取っておく。
・例えば、図Aは反応開始後5時間以降のデータが無い。データを取っていないと図Bで示される様に目的物の最高収率と不純物の生成状況が解らずどの時点で反応を停止させて良いか不明となる。不純物1がある閾値を超えると除去できない場合、最高収率を与える反応時間より前に反応を停止させる必要がある。

・反応時間は分析時間を考慮し、反応速度が速い場合並びに不純物の発生が速い場合は1時間毎、或いは反応速度が遅い場合は1, 2, 4,
8, 16, 24, 36----hr毎にデータ取りを行う(図C)。反応終了時間付近では、必要に応じて試験を行い純度・収量・不純物量・不純物プロファイル確認し、品質規格から最も収率がよく精製可能な不純物量から反応停止条件を決定する。また、反応の進行が時として遅くなる場合があるため、反応条件下で反応時間を延長し反応液中の生成物等の安定性データとのを取っておく。
・反応終了確認は、変換率(原料の残存量)、純度、不純物量及び不純物プロファイルから反応終了規格を設定し、反応はHPLC試験で反応終了規格に従い反応を停止させる。
実験室での反応終了時間が5時間程度の場合

・反応終了確認試験(HPLC)の分析時間が1時間を要すると、その結果はサンプリング時である1時間前の反応状態を見ている。
・実験室では、HPLCが近くにあるためサンプリングから後処理、注入まで短時間で終了させられるが、製造時分析機器まで距離があるためHPLCへの注入に時間が掛かる。HPLC分析時間は45分間以内に終了する分析条件を作製する。
|
反応終了確認は工程内規格に従い実施する。
・反応をリアルタイムで監視できない場合は、効率的なデータ取りを考える。
・スケールアップすると反応の最終段階では原料等の絶対濃度が低下しプロセス開発時より反応速度が遅くなる。
・反応がスムーズに進行するスケールメリット或いは進行しないとスケールデメリットが現れる場合がある。この理由は、撹拌効率等が考えられるが不明なことが多い。
・反応条件下での生成物(目的物)の長時間熱安定性及び不純物プロファイル等の変化を確認し、品質がプロセス開発時と同等かそれ以上かを検証する。
製造時のサンプリング例
・製造現場と試験室が離れている場合、サンプリングから分析までの時間が実験室と異なる(反応は進んでいる)。
 
・製造時は、製造現場と試験室が離れている場合があることから、サンプルから試験開始までの時間が長くなるため試験終了時間とサンプリング時間が交差しない様に試験時間を考える必要がある。
製造現場での反応終了時間が5時間程度の場合
製造現場と試験室との距離があるため、サンプリングポイントと試験終了時間が重ならないHPLC試験時間条件を設定する。

・反応停止(終了)基準に従い実験室と同様に反応の進行チェックを実施する。実験室で最適化した操作法及び品質規格に従い製造現場の実機でも目的物の純度(不純物量、並びに不純物プロファイル)と収率得られるかを検証する。
・医薬品原薬の製造初期は、標準操作法に問題が生じることが多々ある。その時は、製造現場で是正することになるがGMPに従い対処する必 要がある。従って、実験室で発生しそうな操作・条件については、リスクマネージメントが必要となる。そのために、リスクアセスメントを実施し問題点をあぶり出し対処法を持っておくべきである。
|
||
6.反応停止
|
酸・アルカリ・水等の添加、或いは冷却
|
|||
冷却
|
・反応停止剤の滴下或いは冷却時間は短く、反応を短時間で停止できる。
・スケールに合わせた滴下時間の予測と冷却能力から冷却時間を予測し、予測時間の1.5~2倍の時間HPLC分析により熱安定性データ取りと純度・不純物プロファイル等分解のないことを確認する。
|
・反応停止剤の滴下或いは反応温度の降温に長い時間を要するため反応停止に時間を要する。
・パイロット製造で、予測した冷却時間の正確性を確認すると共に、反応停止操作で分解等が無いことを検証し、実製造で実証する。
|
||
滴下
|
実験室での滴下方法:滴下ロート若しくはシリンジポンプ等 ・反応停止剤の滴下は容量が少ないため短時間で済む
 ・滴下時間は、反応時の発熱等で延長することがあり、純度・不純物プロファイルに影響を与える可能性がある。
・プロセス開発時の滴下時間を計測と反応熱が激しいか、緩やかかを確認する。
・反応熱が激しい時は発熱量を計測し(RC-1等)、製造スケール時の予測滴下時間を予測する。
・実験室での滴下時間から実機製造時の滴下時間を予測し、安全率を掛けた時間の熱安定性を担保する。
|
・製造現場では、スケールに合わせ反応停止剤の滴下量が多くなるため長時間を要する。 ・滴下時間の延長は反応停止中に生成物の分解或いは不純物プロファイルの違い等が生じる可能性がある。
・プロセス開発時に最適化した滴下時間或いは熱安定性が担保された 滴下時間を忠実に再現する。
製造現場での滴下方法:滴下槽若しくはダイヤフラムポンプ等

・冷却効率と熱安定性が担保出来た時間内に滴下する方法を考える。
|
||
発熱
|
・反応停止剤の容量が少ないため滴下時の発熱小さいが、発熱による温度上昇を確認して置く。
・反応停止時に生成物等の品質(純度・不純物プロファイル等)に影響を与えない上限温度と時間の熱安定性を確認する。
・出来れば、実機の冷却能力から滴下時の時間を予測し、予測時間の1.5~2倍の時間で生成物の品質確認のデータ取りを行う。
|
・反応停止剤の容量が多く、伝熱面積が小さいため滴下時に発熱大きくなり、時間を要する。
・実験室での発熱量が制御できるか実機のユーティリティーの性能から確認する(予測計算)。
・プロセス開発時に十分な熱安定性のデータがあれば、安心して反応停止できる。
|
||
7.抽出
|
実験室では、分液操作は分液ロートに反応液、抽出溶媒及び水(若しくは、酸或いはアルカリ水溶液)を加え、激しく振り混ぜ静置し分液ロート内で実施する。
 ・分液ロートの場合、抽出効率は抽出溶媒の種類と量と振とう方法並びに回数に左右される。 |
製造現場では、抽出操作は反応を停止させ、抽出溶媒及び水(若しくは、酸或いはアルカリ水溶液)を加え、激しく撹拌後静置し反応槽内で実施する。

・抽出はプロセス開発で設定した溶媒と量及び回数で実施する。抽出効率は撹拌状態と回数に左右されるため分液ロートより高い。
|
||
撹拌
|
・抽出効率は2層間の混合状態に左右される。より細かな滴々を作ることにより抽出表面積が広がり上がる。
・分液ロートは8の字を書くように振れと言われた。激しく振る程抽出効率は上がるが、エマルジョンが発生する可能性が出てくる。
|
・実機では、撹拌翼を最大回転数(或いは、80%程度)で撹拌するため、実験室より抽出効率が良いと言われている。
・実験室でエマルジョンの発生が認められる場合、スケールが上がるとエマルジョン部分は多くなり分離に時間が掛かることが多い。
|
||
時間
|
・一般的に、実験室での2層分離は短時間で終わる。
・エマルジョンが発生してもほとんどの場合は、短時間に2層に分離する可能性がある。
・分液操作でエマルジョンが発生した場合、製造現場では分離に時間が掛かるため一夜放置する場合がある。
・分液状態での熱安定性確認データ取りとして、1, 2, 4,
8, 12, 24, 48~hrのデータ取りを行い抽出溶液中での安定性を確認する。

・エマルジョンを解消する方法として、ろ過或いは少量のアルコールを加える等 がある。
|
・実験室でエマルジョンが発生する場合、スケールアップすると2層の分離に時間が掛かるため一夜静置、或いは週末に静置する場合がある。
・抽出時の生成物の熱安定性を確認していると万が一静置することになっても品質に影響しないため安心できる。逸脱等を未然に防ぐことが出来る。
・製造現場ではエマルジョンが発生した場合、澄明下層・上層を別々に抜き、エマルジョン部分を静置し分離させる。これを繰り返し分離することのより、時間の短縮を図ることがある。

・製造現場でもエマルジョンの解消として、ろ過を実施する場合があるがプロセス開発時にデータ取りと標準操作法に設定していないと逸脱となる。 |
||
8.濃縮
|
実験室での濃縮:ロータリーエバポレーター

・実験室での濃縮は、減圧下ロータリーエバポレーターで実施する。内温は測定できないので、濃縮温度は外温管理となる。
・ロータリーエバポレーターはガラス製であるため、揮発性の酸或いは塩基をそのまま濃縮できる。製造実機では材質の関係で実施できない場合があるので、スケールアップ時は注意を要する。
|
製造現場での濃縮:反応槽(若しくは濃縮槽 )

・製造現場での濃縮は、減圧下反応槽或いは濃縮槽で実施される。反応槽は内温が測定できるが、濃縮温度は外温(ジャケット温度)管理となる。
・生成物等の安定性が悪い場合は、内温管理或いは薄膜式濃縮器(エバポール等)を用いることもある。
・液性により、濃縮槽(反応槽)及び熱交換機の材質を選択する必要がある。酸性ガスが発生せる場合はカーボン製熱交換機を用いる。
|
||
時間
|
・濃縮時間は、溶媒の沸点、容量、外温、減圧度及び回転速度で決まるため、濃縮量が2Lの場合、濃縮時間は~1hr(2L/hr)程度である。
・製造時の抽出溶媒量と濃縮槽の濃縮能力から濃縮時間を予測し、熱安定性のデータ取りを実施する。
・濃縮時の外温と濃縮時間に比例して抽出混合物の熱安定性に影響を与えるため、スケールアップ後の予測濃縮容量と濃縮能力から時間を予測し、その予測時間(1.5~2倍程度の時間)と外温との間の熱安定性を確認し、可能濃縮時間を担保する。
|
・濃縮時間は、溶媒の沸点、容量、外温、減圧度及び撹拌速度で決まる。
・濃縮能力(単位時間当たりの濃縮量)は~200 L/hr程度であるが、各濃縮(反応)槽の減圧度とジャケット温度により異なる。
・例えば、濃縮能力が100~200L/hrで、濃縮容量が2000Lの場合、濃縮時間は10~20hrとなる。
・製造では濃縮時間が増大するため、濃縮中の熱ストレスが反応生成物、類縁体(不純物)等に影響し、生成物の純度が濃縮前後で異なる場合がある。
・従って、熱安定性が担保出来た時間内に濃縮が終了する設備を選択する。
|
||
9.晶析
|
実験室の一般的晶析実験槽
・実験室での晶析条件検討は、一般的に丸底フラスコ-半月板実施されているが、晶析条件の最適化時或いは最適条件の検証に実機により近い幾何学的相似形晶析槽を用いることを推奨する。

・晶析(再結晶)で重要なことは、高回収率、高精製効果(品質規格適合)で原薬(目的物)を得ることが出来、結晶形、粒子径及び結晶癖を揃えることである。
晶析検討用機器
・晶析条件である晶析溶媒、結晶形等の一次検討に96穴プレート、試験管とうにより、晶析(冷却)方法、溶媒量の二次検討にはパラレル合成機等を用い効率的に行う。
 |
製造現場の一般的晶析槽(図面)
・一般的に、製造現場の晶析槽の形状は反応槽と変わりないが、撹拌 翼の形状はアンカー型である。

・晶析液の液性により晶析槽の材質を選択する。
・原薬の結晶粒子径を合わせるためにプロセス開発で設定した撹拌数を製造で使用予定の晶析槽のサイズに合わせて計算する。
・プロセス開発で幾何学的相似形晶析槽を用いて最終設定した晶析の操作条件を正確にトレースする。
・プロセス開発で設定された晶析条件である冷却速度及び撹拌数が実機晶析槽で再現できるかを設備・機器の性能から、或いは実際にそ操作して確認する。
|
||
冷却速度
|
・冷却速度は結晶多形及び結晶サイズに影響を与える。
・正確に降温するためには、自動冷却水循環装置等を使用し、品質目標の結晶サイズに適合するように冷却勾配を求める。
 ・冷却速度(冷却勾配)は出来るだけ製造現場で冷却勾配を再現で きるように「2℃/分、或いは5℃/5分」の様に具体的に設定し、標準 製造法へ記載する。 |
・プロセス開発で最適化した冷却速度を実機晶析槽でも忠実に再現する。
・製造現場で自動温度コントロール(冷却)設備があればセットすればよいが、手動式設備の場合に標準製造法の晶析工程等の冷却指示が「40分で80℃から0℃へ冷却する」では、図Aに示す様に冷却理想曲線である青線でなく、赤及び黒点線の冷却カーブで操作しても逸脱にならない。しかしながら、品質規格の結晶サイズが不適になる可能性がある。図Bの様に晶析時の冷却勾配は「1分間で2℃の割合(2℃/分)で冷却する」とかより具体的に指示することにより冷却理想曲線に近づけることが出来る。
 |
||
撹拌
|
・撹拌状態(撹拌数)は晶析槽・撹拌翼及び邪魔板(バッフル)の形状が違えば異なる。これらの差が結晶粒子径等に影響を与える。
・実機のミニチュア機である幾何学的相似形晶析槽を使用してプロセス開発を実施する。
実機の幾何学的相似形晶析槽(例)
 する。最適化された回転数を使用予定の晶析槽のサイズに合わせて 予測計算する。 |
・プロセス開発で最適化した粒子径、結晶多形等を製造現場で再現するために、実験室の撹拌状態を製造現場で再現させる。
製造で使用予定の実機晶析機(例)

・幾何学的相似形を用いている場合は、撹拌状態のスケールアップは実験機の撹拌数と撹拌翼径と実機サイズの撹拌翼径から次式
Pv = N X D2/3
一定で撹拌数を計算する。
(単位体積当たりの撹拌動力一定の式)
|
||
熟成温度・
時間
|
・晶析の終了は、品質、不純物量と不純物プロファイルの品質規格に適合させるため、並びに収率を確保するために、熟成温度と晶析率規格を設定する。
|
・製造現場では、理由が不明な晶析率が上がらない現象が起こるときがある。
製造現場での晶析の終了は、品質規格に従い晶析率と品質をHPLC試験し決定する。
|
||
ろ 過
|
吸引ろ過、加圧濾過等
・実験室では、ろ過はヌッチェとろ紙で実施されることが多いが、小型の遠心分離機があるので利用できるのであれば最終確認して置く。

・ろ過時のデータはろ過時間を取得する。
・供給液側の圧力p1とろ液側の圧力p2との差圧が同じ時、スケールアップ時のろ過時間はろ過面積に比例する。
・簡易的には、ろ過面積でろ過時間を予測できる。
・ろ過予測時間から、ろ過操作が2日に渡る可能性があるため、目的物の熱安定性等のデータ取りを取って置く。
|
遠心分離機(縦型・横型)、ろ過乾燥機、加圧濾過器等
・製造現でのろ過は、遠心分離機を用いることが多いが、酸素等の影響を受ける場合は加圧濾過器を用いる。

・ろ過予測時間から目的物の熱安定性等を考慮し、実験室のデータから得られたろ過物及び晶析槽内の結晶の保管条件を考慮する。
|
||
10.乾燥
|
コルベン式真空乾燥・棚式真空乾燥・棚式送風乾燥機等
実験室の乾燥機(棚式真空乾燥機)
・乾燥機の選択はろ過後の湿晶の状態から適した乾燥機を選択する。棚式真空乾燥機が万能型乾燥機として一般的に用いられる。
 ・乾燥条件の設定では、熱に安定或いは不安定な場合、並びに酸素に 不安定な場合等に合わせて乾燥機の選択或いは条件を最適化する必 要がある。
・目的物である中間体・原薬・不純物等の熱安定性では乾燥温度と乾燥時間が問題となる。それらが分解しない温度と時間の熱安定性データを取得する必要がある。
・乾燥機は、ろ過後の湿晶が大きく流動性がある場合は各種乾燥機が適用できるが、湿晶がwet率の高いケーキ状態である場合は棚式乾燥機、湿晶が酸素或いは熱に弱い場合は加圧ろ過乾燥機等を選択する。
・コニカル真空乾燥機は回転するため得られた結晶を均一に混合することが出来が、湿晶のwet率が高いケーキ状態等では塊(ダマ)になる可能性がある。選定に当たっては、実験室で湿晶をコルベンに入れエバポレーターで加熱減圧下回転数を変化させながら塊にならない回転条件を確認する必要がある。
 ・乾燥終了確認は、原薬の品質規格(水分値、溶媒の含量等)に従い設定した工程内規格値及び試験方法により実施する。 |
コルベン式真空乾燥・棚式真空乾燥・棚式送風乾燥機・コニカル乾燥・振動乾燥機・ろ過乾燥機等
製造現場の乾燥機
・湿晶の熱安定性が確保されている場合は、棚式乾燥機或いはコニカル乾燥機を用いる。熱・酸素等に不安定な場合は加圧ろ過機等を用いて窒素等で圧を掛けろ過し加熱窒素或いは加湿窒素を通気して溶媒を飛ばす方法をとる。加湿窒素は水和物をコントロールする時にも用いられる。

・棚式真空乾燥機は汎用性が広く湿晶の乾燥に一般的に用いられる。微細でwet率の高い湿晶の場合は乾燥後に塊となる可能性があるが、結晶の取り出しが容易であるため塊を崩すことが可能である。
・コニカル乾燥機は連続或いは間欠回転、並びに一定の時間で回転数等を設定することが出来る。実験室で得たデータに従い回転数/時間を決定し、パイロット製造時に検証する。
・乾燥晶が大きな塊になる場合、グラスライニング製コニカル乾燥機を損傷或いは取り出せない可能性が出て来る。
・乾燥時間は湿晶の状態と量、並びに乾燥機の大きさと能力により影響されるため設定しない。乾燥終了確認は標準製造法の工程内規格に従い判定する 。
加圧ろ過機と加湿窒素
・酸素或いは熱に弱い中間体或いは原薬の場合は、湿晶の乾燥に加圧ろ過機を用いて窒素加圧によりろ過し加湿窒素、乾燥窒素或いは加熱窒素を通過させ乾燥させる。また、加湿窒素は結晶水和物の制御にも有用である。
 | ||

適合させる各工程の最適操作条件を確定させるために実験室で繰り返し原薬を合成し、実験室での原料・副原料・試薬・生成物・類縁体(不純物)等の熱安定性を担保している。しかしながら、殆どスケールに合わせた操作時間を考慮した熱安定性データを取得していない。
-実験室の最適操作条件を製造現場で忠実に再現するために-
最適化
項目
|
検討(データ取り)項目
|
備考
|
|
原料・副原料・試薬等の選定と品質
|
・原薬の製造に当たって、出発物質は当局が認める承認申請上必要な工程数を考慮して原薬の合成ルート中の中間体から選定する。
・出発物質が決まっている場合は変更しない。
・出発物質が決まっていない場合は、上記に述べた様に、ICHのガイドライン及び当局の規制に従い選定する。
・原薬の目標品質を設定する。
・出発物質は原薬の目標品質に適合出来る品質でなければならない。
・また、原料(出発物質を含む)・副原料・試薬・触媒及び溶媒等も原薬の目標品質に適合出来無ければならない
・副原料・試薬・触媒及び溶媒等は、大量に入手可能な化学薬品を採用する。
・使用する原料(出発物質を含む)・副原料・試薬・触媒及び溶媒等の化学物質は、毒性試験・SDS等により作業者への安全性、環境への負荷並びに危険性を評価すべきである。
|
・出発物質(原料)の選定は、原薬の品質、GMP管理、コスト等に影響を与えるので重要である。
・原料(出発物質を含む)・副原料・試薬・触媒及び溶媒等は品質規格及び試験方法を確認する。試験方法が無い場合は、規格及び試験方法を作成する必要がある。
・出発物質はTLC, IR と NMRで確認試験を実施された後、HPLCによる純度試験(但し、含まれる総ての化合物が分析出来るHPLC条件でなければならない)により純度と不純物のプロファイルの分析を実施する必要がある。
・出発物質(原料)の品質確認は、製造に使用するロットを用いて原薬が目標品質に適合するかユースエストを確認する。
・SDSの調査で安全性及び有害性が確認された場合は、化学物質の取り扱い、仕込方法等を細かく取り決める必要がある。
|
|
反応容器等の形状・材質
|
・一般的に実験室では、反応はガラス製丸底フラスコを用いてデータ取りされているが、スケールアップ前には製造実機を反映したガラス製幾何学的相似形反応槽を用いで操作中の状況の確認と最適化条件の検証を実施されるべきである。
・反応条件の液性(酸性・塩基性・粘性等)或いは反応条件により製造設備の材質(SUS,
GL, ハステロイ、樹脂製等)や撹拌翼等の形状の検討と選択を行う。
・フッ素化合物の反応等では、GL製反応槽の表面が腐食されることがあるので注意を要する。

・幾何学的相似形反応装置は、実機反応槽の設計図があれば撹拌翼、邪魔板(バッフル)等をスケールダウンして作製することが出来る。また、装置の底に取り出し用のコックも付けることが出来る。
|
・現在のGL(グラスライニング製)反応槽は耐アルカリ性が高くなっているが、液性がアルカリの場合はSUS(ステンレススチール)反応槽を、酸性の場合はGL製反応槽を用いる。
・極低温反応の場合には、SUS・ハステロイ等の金属性の反応槽は冷却効率が高いことから選択されるべき である。
・超低温反応には、冷却効果が高い槽内に蛇管式ジャケットを持った反応槽等を用いる
・スケールアップ前には、反応は現場実機の幾何学的相似形反応槽を用いて撹拌状態(撹拌数・撹拌翼の形状を含め)・反応速度・不純物量・操作条件等の相似性の検証されるべきである。
 |
|
仕込み
方法
|
・仕込み(投入)は、原材料を直接投入、反応溶媒等に溶解した溶液の投入、ライン投入、減圧吸引、或いは圧送移送等の中から最適な投入方法、仕込み速度、仕込み時温度並びに順番等を検討する。
・原料・副原料・試薬・触媒・溶媒等(原材料)の危険性及び有害の評価と無水反応、酸素ケアが必要な反応での不活性ガス等の必要性を確認すると共に仕込み方法を検討する。
・原材料の仕込み温度、時間及び順番が危険性(反応の暴走)・安全性(作業者への有害性)・安定性(目的物の品質)等に与える影響を検証する。
 |
・仕込み・反応・その他の操作について、原料等の危険性・安全性をSDS、危険物ハンドブック等で一次評価を実施する。
・使用物質の可燃性、静電気、危険性、有害性等から安全対策と仕込み方法(オープン或いはクローズド)を決定する。
・仕込み(方法・温度・時間・安全性・順番等)操作が実際に製造現場で可能かどうかを検証する。
・検討した仕込み方法が不可の場合は、工夫をするか、代替え案を考える必要がある。
 |
|
反応溶媒
|
・反応溶媒は反応進行(イオン反応:SN1,
SN2、ラジカル反応等)に影響を与えるため、反応効果・品質(純度、不純物量・不純物プロファイル等)・残留溶媒ガイドラインのクラス・収率並びに危険性等を考慮し選択する。
反応溶媒種の選定
・反応溶媒は、類似の反応条件或いは委託先の指定から選択することになる。迅速に反応溶媒と反応濃度(溶媒量比)のスクリーニングを行うために、コルベンを並べて行うか、パラレル合成機等を用いて効率よく検討する。
・反応進行確認は迅速に一斉に判定するために TLC等を用いて行うことを薦める。しかしながら、TLCは定性的判定となるので反応の進行と最適な反応溶媒の絞り込に用いる。

・一次スクリーニングで反応溶媒が何種類かに選定されたなら二次選定として、それらの溶媒を用いて再現した反応液、或いは一次スクリーニングの反応処理液を用いて定量化できるHPLCで分析する。二次選定として反応速度、目的物の品質(純度、不純物量、不純物プロファイル)及び収率(変換率)により最適な反応溶媒を決定する。
・反応溶媒の選定は、HPLCにより反応生成物の純度、不純物量、不純物プロファイル、変換率及び反応速度等から行うが、商業生産では大量に溶媒を使用することから回収精製の容易さ・コスト等で選定することも考慮する。
・反応溶媒等は残留溶媒のガイドラインのなるべくクラス 3 かクラス 2 を選択する。
|
・反応溶媒等は反応効果(反応時間、収率、純度、不純物プロファイル等)、抽出溶媒、静電気等の安全対 策などを考慮して選択されるべきである。
・原薬の製造コストは重要であり、製造効率を上げる溶媒種と量を設定されるべきである
・反応条件の選定の一時スクリーニングには、迅速かつ一斉に分析できる
TLC法 を推奨する。
・HPLC は、分析時間に30~60分程度要し、反応条件の初期設定から使用するとプロセス開発に時間が掛かりすぎ効率が悪い。但し、HPLC法は定量的に扱えるため、反応溶媒の最適化と最終決定する場合は用いるべきである
定量的反応チェック(HPLC)
 
・反応溶媒と抽出溶媒等操作に使用する溶媒はなるべく単一の溶媒とし、回収精製再使用が容易で反応後処理・濃縮等に安定で、濃縮時に共沸脱水しやすい溶媒を選択する。
|
|
反応条件
|
・反応条件の最適化として、原料と副原料・試薬・触媒等の仕込み量比、仕込みの順番、副原料・試薬の投入(滴下)方法と速度、反応溶媒の種類と量(反応濃度)、反応温度・圧力及び撹拌(数)状態等の操作条件を最適化する。
・投入方法では、固体、液体並びに製造スケールにより変更させる必要性が生じる。
・投入方法・投入時間、投入時の温度と原料・生成物等の安定性データ取りと投入方法を決定する。
実験室での液体の投入・滴下方法

・反応条件等の最適化は、標準操作法が在るか無いかで大きく異なる。
・標準操作法がない場合は、化学反応データベース(SciFinder,
Reaxys等)から類似の反応を検索し、その反応条件を基本として反応(操作)条件の最適化する必要がある。
・反応操作条件の検討は、最適値を見出すことであるが、その最適値に安全域を確保させ品質に影響尾を与えない操作条件の範囲を設定するためにデータ取りを実施する。
・委託先からの標準操作法等がある場合は、その反応条件を基本とし、安全域の確認と操作範囲の決定が主なデータ取りとなる。
・反応条件の最適化は多岐にわたる条件検討が必要となるため、溶媒種の選定と同様に下記に示すようにパラレル反応機等を用いて効率的に実施する。
反応条件となる原料(出発物質)と副原料・試薬・触媒等の仕込み量比及び反応溶媒の種類と量比
・創薬或いは委託先からの反応条件がある場合は、その反応条件(温度、試薬・副原料・塩基等の量比等)を基本的に採用する。
・原料(出発物質)と副原料・試薬・触媒等の仕込み量比及び反応溶媒濃度は、溶媒の選定と同様にパラレル反応機を用い、原料に対して副原料・試薬・触媒等を一次検討として0.5等量刻みに変量し反応進行状況を確認する。次に二次検討として反応条件の最適値近くでは、0.05~0.1等量刻みで最適値を求める。最適値を目標値(操作管理値)として、目的物が品質目標に適合する反応条件を目標値を中心に操作(管理)範囲を定める。この時、反応の進行状況はHPLC試験により確認する。
副原料の量比
・原料 (1.00) に対して副原料・試薬等の量比(化学当量)の最適化は、量比を0.1或いは0.2当量ずつ変量し最適範囲を求め、その範囲付近では0.01~0.02当量で細かく変量して最適値を求める。

触媒の量比
・触媒量は、金属触媒は非常に高価であるため、反応の速度、目的物の純度及び変換率により少ない最適量を求める。例えば、触媒5%或いは10% Pd/Cでは原料と触媒の重量比10 %以下とし、重量比10%から0.2%ずつ減量して最適範囲を求め、更に細かく変量して最適値を求める。
・重量比でPd/Cの10%で反応が進行しない或いは非常に遅い場合は、増量して行く。より高価な金属触媒(Rh,
Ru, Pt等)では触媒コストに応じて0.01
mol%以下で最適化し、最適値を求める。
・酸、塩基等の触媒量では、重量比若しくはモル比で最適値を求める。
・原料と試薬・触媒等の量比は最適値を求めてデータ取りされるが、それらの許容範囲と標準製造法の設定値(最適値)も同様に確認して置く。
反応濃度、仕込み順番及び投入速度
・反応液濃度は、創薬研究或いは委託先からの指定を用いる。
・濃度の最適化に関しては、1 mol/Lを基本とし、品質(純度、不純物量、不純物プロファイル等)、反応速度、変換率と仕込効率等の経済性を考慮して最適濃度を決める。
・副原料・試薬等の仕込みの順番は、反応の暴走或いは不純物の増加につながるため、検討を要する。
・反応速度が速い場合、副原料・試薬等の投入(滴下)速度に影響するため、滴下速度は反応混合物の昇温と反応速度の状況を確認しながら設定する。
・これらの設定はHPLC試験により確認し、目的物の純度、変換率から設定する。
反応温度(圧力等)の最適化と設定
・指定された反応温度がある場合は、指定(目標)温度を中心に最低±10℃
の安全域を確認する。
・例えば、指定された反応温度(例えば、目標温度80℃)を中心に±10℃での反応時間並びに中間体・原薬等の品質目標(製品・中間体規格)及び期待収量(変換率)が適合するかを検証する。問題ない場合は、反応条件許容範囲となり、80±5℃が操作管理範囲とする。
・反応温度が指定されていない場合は、類似の反応例から反応温度(例えば、80℃)を選択し、±10℃刻みで反応進行状況を確認する。反応進行状況は反応時間並びに中間体・原薬等の品質目標(製品・中間体規格)及び期待収量(変換率)に適合するかを検証する。
・反応温度80℃の反応進行状況が最も良ければ、±2℃刻み(或いは±5℃刻み)でデータ取りと反応時間並びに中間体・原薬等の品質目標(製品・中間体規格)及び収量(変換率)から最適化する。

・反応温度の設定は反応温度と反応時時間による中間体・原薬等の品質、変換率、不純物量と不純物プロファイルから品質目標(製品・中間体規格)及び収量から最適温度(目標温度、例えば82℃)と許容操作範囲(例えば、70~90℃)と操作(管理)範囲(例えば、77~87℃)を設定する。

・反応温度は恒常的に原薬が品質目標(純度・不純物量・不純物プロファイル等)に適合出来、最高収率で得られる様に設定する。
・仕込み、反応及び後処理温度(圧力)等の反応条件は範囲で設定する。
・その範囲に操作許容範囲(中間体・原薬の品質規格に適合する操作条件範囲)を決める。
・操作許容範囲に安全域を取った操作範囲及び実際に運転する目標値を設定する。例えば、反応温度に最低±10℃の安全域を取り設定する。
・次に、その操作範囲に実際に製造現場で操作する目標値を設定する。
HPLC法による反応のチェック(例)
・HPLC法では1機器1反応液しか分析出来ないが、分析結果を定量的に取り扱えるため、プロセス開発での条件の最適化に有用である。
・以上のようにして設定した反応条件による反応の進行はHPLC試験によって行われる。
・反応チェックは基本的には 1 時間毎にサンプリングして実施するが、反応時間が不明な場合は1,
2, 4, 8, 12, 24----時間等で実施する。
・反応時間は条件の最適化時にTLC分析を実施しているのでおよその反応時間が解っていることが多い。その場合は反応終了時間の1~2時間前から終了後~2時間程度まで1時間毎に行う。
実験室でのサンプリング例
反応時間が短い(10時間まで)場合

反応時間が長い(12時間以上)、或いは不明な場合

反応終了時間が5時間程度と分かっている場合は、反応後4時間目から1時間毎に、また7~8時間までサンプリングして反応混合物の変化の推移を確認する。反応条件下での時間と反応混合物の安定性を確認する。
・反応終了確認後もHPLCで分析し、目的物、不純物等の変化推移を担保して置けば、製造時に逸脱があっても担保している時間内であれば品質を保証できる。
・HPLC分析の結果は、~1時間前の反応状態を見ていることから、反応終了確認後最低2時間以上撹拌続行し、熱安定性による品質確認を行う。
・仕込み時、反応中等の温度推移(発熱・吸熱)を記録・確認し、スケールアップ時の発熱量を予測しその対応を考える。
 |
・反応条件の最適化のデータ取りには、一変量実験法(従来の方法)と多変量実験法(より進んだ方法)があるが、デザインスペースの考え方から、最近では多変量実験による多変量解析へ移りつつある。
・多変量解析法の理解が乏しいので省略する。
・化学反応は、温度・濃度・圧力・光・触媒の種類と量・反応種の表面積等により影響を受ける。
・反応条件は高純度で、高変換率で、不純物の極めて少ない目的物を与える様に最適化させる。
・実験室で最適化した反応条件はスケールアップ後の製造実機で忠実に再現出来なければ、実験室と同等品質の原薬は得られない。しかしながら、忠実に再現させることは不可能に近い。
・実験室で最適化した反応条件を製造現場で再現させるためには、最適化した製造スケールに合わせた条件に組み直す必要がある。そのためには、実験室でのスケールアップシミュレーションが正確に実施できるかが非常に重要となる。
・組み直した反応(操作)条件の最適化は実験室⇆パイロットプラント⇆プラントの繰り返しにより修正して行く。
製造現場での液体の投入・滴下方法
・実験室の投入時の条件を再現するためには、投入時間、投入時の温度と原料・生成物等の安定性が問題となるため、スケールに合わせた投入方法を考える。
・滴下ロートのスケールアップでは、ダイヤフラムポンプ或いは滴下槽等の代替えを考える。
・反応条件の最適化は、一変量解析として1ファクターのみを変量させる方法でデータ取りしていたが、現在では、多変量解析が進み複数ファクターを一度に変量させ反応条件検討を効率的に進める方法が主流になって来ている。
反応因子の変量検討例

反応条件として、触媒種の検討を反応温度120℃で実施し、触媒として硫酸水素ナトリウムが最も良い収率を与えた。

硫酸水素ナトリウムを触媒として、原料(phytosterol)と不飽和脂肪酸エステル比、触媒量、反応時間及び反応温度の最適化の実施例を示している。
 
・以上の様にして得た実験室で設定反応条件を製造現場で忠実に再現すること。
・水素添加触媒であるPd/Cはdryであり大量に取り扱う場合、発火する可能性が増大ある。非常に危険であるため50%
wet Pd/Cを用いるべきである。
・製造現場では、実験室で設定した反応温度(全ての操作条件を含む)は幅で管理するが、目標値(最適値)の温度(条件)で操作するように指示する。
・温度範囲内の目標値は反応促進に最適な反応(操作)条件であるこ と。
・反応(操作)温度範囲及び実際に運転する目標温度を維持できるユーティリティーと製造設備・機器能力の確認と選定を行う。
・製造設備・機器が操作条件を維持・反映できないことが判明した場合は、設備等の改造を考える。
スケールアップ時に変更してはいけない数値と変更すべき数値がある。

・反応濃度は、1 mol/Lを基本とすることを推奨するが、反応混合物の粘度、撹拌状態、反応速度、発熱状況並びに分子内反応等から決定する。
・反応温度(全ての操作温度)は製造現場で±3℃以内であれば自動は勿論のことに手動操作でも製造現場の担当者により制御できる。
・反応温度(条件)は高品質・高収率だけでなく、経済面から反応速度(時間)等を考慮して決定することも許容される。
・反応温度の許容範囲が広く取れれば、不慮の出来事が生じ温度操作範囲が逸脱しても品質への影響が担保出来ていれば中間体・原薬の品質を保証することができる。
・反応条件のワーストケースで実施した時に、品質及び収率へ影響を与えた場合は反応条件の設定範囲を狭めると共に、製造実機の性能・能力等が設定範囲を再現できるかの検証が必要である。
操作の重要変動要因(パラメータ:温度・pH・圧力等)条件の設定基準
・データ取りで求めた最適値を標準操作法の管理目標値とし、品質目標に適合する操作条件を目標値を中心に操作管理範囲として設定する。操作管理範囲は標準操作法の操作範囲とするが、現場での運転は出来る限り目標値±2℃以内で運転(管理)する。

TLC法
・分析は反応の一次スクリーニングにも使用することが出来るが、定量化できないため反応条件の絞り込みには向かないかもしれない。

・しかし、TLCは全ての有機物を一斉に分析でき、反応条件検討の初期の段階では反応速度(反応時間)を簡易的に把握でき、HPLC分析の補助としても有用である。
HPLC法
・反応条件の最適化は、HPLC(UPLC)を用いて反応チェックを実施し、純度、変換率、不純物量等を数値化し、品質目標に適合する最適な反応条件を設定することになる。

・但し、HPLC分析は反応液に含まれる総ての不純物等を分析出来ているか不明な場合がある。従って、TLCとの組み合わせと、HPLC分析条件(溶出条件)を設定(工程内分析法)することが非常に重要となる。
反応条件のその他の検討項目
・発熱反応では、総発熱量・暴走温度等を確認する。
・実験室での発熱は、小さいかもしれないが、製造量が増加すると製造現場では膨大な発熱になる可能性がある。制御できなければ大惨事となる。
・反応の危険性・安全性は示差走査熱量計(DSC)、反応熱量計(RC-1)、加速速度熱量計(ARC)等で危険性評価し、反応温度の管理方法を設定する。
・反応温度と分解温度が近い場合、或いは激しい発熱反応は極めて危険である。
・反応の危険性(設定操作温度の >+20℃の危険性)を評価する。
・激しい発熱を伴う反応の場合は、冷却による熱管理を検討するより、副原料或いは試薬等の投入方法等により熱管理が出来ないかを考える。
反応条件の最終最適化
・反応条件の最終最適化は、幾何学的相似形反応機(実機のミニチュア機)を用いてスケールダウンシミュレーション及び/或いはスケールアップシミュレーションを実施し決定することを薦める。
・また、反応等の設定(最適化)した操作範囲(温度・撹拌状態・操作時間等の条件)のワーストケースでスケールダウンシミュレーションを実施し、目的物(中間体或いは原薬)の品質・収率等に与える影響を検証する。

反応条件が最適化された塩酸リトドリンの先発品のHPLC チャート
 |
|
撹
拌 状 態 |
・反応中の撹拌状態(撹拌数)は反応進行、反応時間、純度、収量及び不純物プロファイル・量等に影響を与える場合がある。
・撹拌状態が反応状態に影響を与えない場合は、実機反応槽の撹拌機の最大撹拌数の80%程度に設定する(余裕を持たせる)。
・撹拌状態が反応に影響を与える場合は、出来る限り幾何学的相似形反応槽を用いて撹拌数と反応進行状況並びに目的物の品質を評価して撹拌数を最適値を求める。
・この時スケールアップ製造に予定している実機反応槽の最大撹拌数を把握し、実験室での最適撹拌数と品質に影響を与えない撹拌数範囲のデータ取りを行う。
・スケールアップは、幾何学的相似形反応槽で得られた最適撹拌数を実機で撹拌状態を同等にするためには、「単位体積当たり( )の撹拌動力(Pv)一定」で撹拌数のスケールアップすることである。

・撹拌数の予測計算値を実機(パイロット、プラント)に適用し、実製造で得られる中間体及び原薬が実験室で得られた中間体と原薬の品質と同等以上の目標品質(純度、不純物量、不純物プロファイル)に適合し、変換率等に適合することを確認し撹拌数を確定させる。
|
・反応時の撹拌は、反応槽内の溶液中の混合物・温度・濃度を斑なく均一にし反応速度を一定にして、異常反応を抑制し目的物の品質(純度、不純物量、不純物プロファイル)を安定させるために行われる。
・実験機の最適撹拌数の実機への適用は、幾何学的相似形反応機で得られた撹拌数からスケールアップ後の反応槽に合わせ、
PV = N X d2/3
N’ (実機撹拌数) = N X d 2/3/d’2/3
N: 実験機撹拌数、N’: 実機撹拌数、d: 実験機槽径、d’: 実機槽径、D: 実験機翼径、D’: 実機翼径

・同じ回転数では、大小の撹拌翼の仕事量が違う。
 |
|
加熱
・
冷却 |
実験室での加熱或いは冷却
・実験室では、大量の熱媒或いは冷媒で反応を含めた操作温度の管理が行われる。また、加熱・冷却方法並びに媒体量が異なることから、実験室でのプロセス開発時のデータを基にスケールアップ時の加熱・冷却に必要な時間と必要な熱媒・冷媒温度を外挿しにくい。
加熱
・加熱は水浴、オイルバス、マントルヒーター等で行う。

冷却
・冷却は冷却機或いはドライアイス-アセトン(メタノール)等により行われる。

加熱・冷却のデータの取得
加熱
・操作温度と熱媒(指定温水、蒸気等)の選択として、熱媒による操作温度への昇温時間による熱安定性データの取得。
・熱安定性データには、スケールアップを想定して予測される昇温時間に安全率を掛けてデータ取りする。
冷却
・反応温度と使用する冷媒温度及び冷却速度の関係、並びに反応温度を維持するために必要な冷媒温度等を取得する。
・反応時間は、製造コストを工程の仕込みから反応後処理までを8時間以内になる反応条件を検討することを推奨する。
反応槽容量による伝熱面積2*

計算に用いている総括伝熱係数3*

・但し、総括伝熱係数は各工場のユーティリティーの性能、反応
槽材質等により異なるので注意が必要である。概算計算では上 記の数 値で問題ない。
|
製造現場での加熱或いは冷却
・製造現場の実機反応槽では、薄い幅のジャケット内へ熱媒或いは冷媒を循環させることにより反応温度を含む操作温度の管理を行ことから、実験室と違い加熱・冷却効率が低い。
・加熱・冷却に時間を要する。また、実機反応槽のスケールアップに於いても容量に対する伝熱面積(ジャケットと反応槽が接触する面積)は、反応槽の容量が10倍増加しても伝熱面積は約5倍程度しか増加しない。
・特に、低温反応では反応温度を保持させることが困難になる場合がある。

・スケールアップ時に加熱・冷却速度、反応温度(特に、低温)を維持するためには、 冷媒温度を下げる必要がある。
・低温反応では、実験室から一機にスケールアップするのでなく、~100
L程度のパイロット機を用いて製造現場のユーティリティーを用いて冷却時のデータを取得することを薦める。
加熱・冷却のデータの取得
・加熱
熱媒(指定温水、蒸気等)の温度と所定の温度への速度
・冷却
反応温度と使用する冷媒温度及び冷却速度の関係、並びに反応温度を維持するために必要な冷媒温度等を取得する。
加熱・冷却時の熱媒・冷媒温度による加熱・冷却時間計算
加熱・冷却到達時間θはか式で計算される。

上記計算式から作成した加熱・冷却到達時間計算表

・上記の計算シートは、スケールアップ時に選択した熱媒での加熱或いは冷却時の到達時間を予測するのに利用することが出来る。
・但し、各工場のユーティリティーの性能、反応槽材質等により総括伝熱係数が異なるので注意が必要そうかつでんえ
2* 反応槽の伝熱面積(八光産業の技術データ):http://www.hakko-sangyo.co.jp/wp-content/uploads/2013/10/ab4b9ed0c028ef45ad95d6ac33f1b132.pdf |
|
反応終了確認
|
・反応終了は、HPLC分析により反応終点規格に適合しているかを確認し、判断する。
・反応終点は目的物の純度・不純物量・不純物プロファイル・変換率等の単独或いは組み合わせで設定される。
・反応終点規格は標準製造法に各工程の工程内規格として制定される。
・反応進行状態の確認は、HPLCで行うが、反応速度が速い場合は1時間毎に、遅い場合は1, 2, 4,
8, (12), 24, 48 hr 等で実施する。

・但し、HPLCの分析結果はサンプリングした時の反応状態を確認しているので、分析終了時の反応進行状態と異なっている。
・HPLC分析結果は30~60分(サンプリングから分析終了の時間)前の反応状態
・HPLC分析に0.5~1時間程度を要する。反応終了確認の分析時間を短縮するためHPLC溶出条件の確立、或はUPLCの使用を薦める。
・このことからも、反応終了確認後も目的物の純度、変換率、不純物量及び不純物プロファイル等を確認することにより、反応時間の進行に伴う状況推移を確認し、反応停止条件が適格か或いは変更が必要かを判断できる。
|
・反応停止は分析後となるため、反応はさらに進行(~1 hr.)していることを考慮する。
・反応の安定性、不純物の増加及び不純物プロファイルに変化がないことを検証するために、反応終了後も同条件下で生成物の安定性を工程内規格を逸脱するまで確認する。
・また、1時間毎にサンプリングするためには、分析時間を40分間以下にする必要がある。
・反応の進行は、反応時間でなく反応終点規格を設定し管理する。
・製造現場では、サンプリングからHPLC分析終了までに時間が掛かることを考慮し分析条件を決定する。
・反応速度が遅い場合或いは反応速度を上げると不純物が増加する場合等は、8時間に拘らず最適条件を求める。
・HPLCの分析条件は反応混合物の全ての化合物(生成物、原料、副原料、試薬、不純物等)が分析出来なければならない。
|
|
後処理・液性
|
・反応停止条件(冷却或いは酸、塩基、水等の添加)の検討と生成物の品質への影響を確認。
・酸或いは塩基、水等の添加は殆ど場合に発熱を伴うため、生成物の熱安定性が問題となることから、反応停止時の発熱量の確認と処理温度の設定。
・反応後処理温度は品質への影響から○○温度以下或いは範囲として設定
・酸・塩基の添加による反応停止終了確認時のpHは試験紙か、pHメーターか?pHメーターの場合は幅で設定し、目標pHを設定
|
・反応停止時の生成物の品質への影響を確認する。操作時間の最低2倍程度の安定性確認
・反応停止時に発熱・分解等が無い最適な酸或いは塩基、水等を選択する
・反応処理上限温度に安全率を掛ける。
・品質への影響を考え選択する。幅で管理する。
|
|
後処理時間・温度
|
・反応後処理での品質への影響は、熱安定性として処理時間と温度範囲(純度・不純物プロファイル)の確認
・処理時間及び温度を設定する。
(熱安定性:○○温度の+5~10℃で、1, 2, 4, 8, (12), 24, 48 hr で確認)
|
・最低±10℃の品質に対する安全域を取る。
・スケールに合わせた反応処理時間
・安全域の最高及び最低温度での後処理時の反応
液の状態を確認
|
|
抽出・ 洗浄
|
・抽出溶媒は溶解度、反応性、次工程の溶媒、共沸脱水効果を考慮して設定
・抽出溶液使用量は溶解度等から~5V(W)/W以内で設定。
・抽出液の洗浄液と方法は安定性並びに物性による精製効果の確認と設定。
|
・容積率、共沸脱水効果、次工程への溶媒置換を考える。
・容積率、コストを考える
・物性が違えば、溶媒の種類、溶解度、pH、吸着、キレート化等で精製できる。
|
|
操作停止
箇所
|
・安定的に操作停止できる箇所の確認と停止箇所及び停止時間の設定。
・保管条件である場所、温度、不活性ガスの有無等の判断 ・設定に当たっては、熱安定性のデータが必要であり、例えば、~5 日間の熱安定性の検討。 |
・各工程は1日では終わらない場合がある。
・また、長期の連休等があるため、出来るだけ長く安定性試験を実施する。 |
|
乾燥剤
|
・脱水操作として、乾燥剤の使用か、或いは共沸かを選択する。
・抽出液の乾燥は一般的には硫酸ナトリウム(芒硝)、硫酸マグネシウムを用いて実施するが、脱水効果は共沸濃縮操作の方が高い。
・共沸操作を選択するためには、(濃縮温度に要する時間+α(安全率))X回 数の熱安定性試験を実施する。 |
・乾燥剤の脱水効果は小さい。
・乾燥剤を使用すると、反応槽、ろ過器等の洗浄が必要となり、コスト及び時間が掛かる。
・共沸脱水操作で得られた濃縮物は無水反応であるグリニヤー反応を実施することが出来る。
|
|
濃縮
|
・濃縮温度と実機濃縮性能の確認と抽出溶媒種と量から濃縮時間を予測。
・濃縮温度は抽出液を濃縮予定温度(最低 +5℃)に加熱し、予測濃縮時間の1.5~2倍時間の熱安定性(純度・不純物プロファイル等)を確認する。目的 (生成物)の熱安定性から濃縮温度を設定する。 ・濃縮温度は熱安定性から、外温40℃以下(例)として設定する。  |
・前もって全ての実機の濃縮槽・反応槽のデータを持って置くと、濃縮時間の予測に有用である。
・濃縮時の熱安定性のデータは、一般的に外温(熱媒温度)で、目的物の純度の経時変化を確認し、濃縮時間の最大時間を確認する。 ・但し、濃縮時間に問題がある場合は、その時間内に濃縮が終了する濃縮設備(薄膜蒸留機等)を検討する。 ・必要に応じて抽出溶媒或いは次工程の反応溶媒での共沸による脱水効果と最終水分値を確認する。 ・次工程の反応溶媒へ置換(品質・不純物プロファイル)等を考える場合は、置換濃縮時の安定性を確認して置く。 |
|
保管
|
・中間体等を安定に保管するためには、温度・期間・湿度等の安定性データが必要となる。
・保管条件の確認は、中間体の予定保管温度(室温保管の場合は、30℃)に+5℃で安定性試験を実施し、安定な期間を確認、設定する。
|
・次工程の溶媒へ置換(品質・不純物プロファイル)時の安定性デートの取得
・保管条件下での安定性は、温度と保管時間に安全域(余裕)を取り保管条件の十分な担保を取る。
|
3* 総括伝熱係数:
http://www.kobelco-eco.co.jp/product/process/docs/glass_tech_1-4.pdf
web上の伝熱計算シート;ジャケット&攪拌機付きタンクの伝熱計算Ver1.1(ライセンスキー必要):
操作
|
条件
|
作業時間
|
仕込
|
固体
|
1 品目(150 Kg)/~0.5 hr.
|
液体
|
1000 L/~0.5 hr.
|
|
溶媒
|
1000 L/~0.5 hr.
|
|
滴下
|
発熱少ない(あり)
|
100 L/~0.5 hr.(~2.0 hr. Lab. データ
から計算)
|
昇温
|
全ての温度
|
0.5 hr. ~
|
冷却
|
100℃⇒25℃
|
0.5 hr. ~
|
25℃⇒0℃
|
0.5 hr. ~
|
|
0℃⇒-30℃
|
1 hr.~
|
|
濃縮
|
減圧下・外温40℃
|
100~200 L/hr.
|
抽出
|
上層を取る
|
0.5 hr.~
|
洗浄
|
0.5 hr.~
|
|
下層を取る(逆抽出)
|
1 hr.~
|
|
ろ過
|
活性炭ろ過
|
0.5 hr.~
|
中和
|
冷却しながら
|
0.5 hr.~
|
晶析
(再結晶)
|
仕込
|
~0.5 hr.
|
加熱溶解
|
~1 hr.
|
|
ろ過
|
~0.5 hr.
|
|
晶析(冷却・熟成)
|
~12 hr.
|
|
遠心分離
|
36 inch
|
50 Kg/~1.0 hr.
|
乾燥
|
乾燥入れから取出しまで
|
8~24 hr
|
スケール
|
評価・検討項目
|
検討部門
|
||
研究
|
~0.1 L
|
○原料(出発物質)、副原料及び試薬等の危険性・安定性
○中間体・原薬の安定性・有害性
|
創薬研究所
|
|
プロセス開発
|
製造法の検討
|
~1.0 L
|
○使用するすべての物質のSDS等による安全性、危 険性と仕込み方法の検討
○危険性・安全性リスクアセスメントの実施計画立 案
○反応時の発熱状況、+20℃の安全性
○加速危険性(DSC、RC-1、ARC等)
|
プロセス(工業化学)研究所
・有機合成化学者
・化学工学者
・分析化学者
|
スケールアップ
(プロセス)検討
|
~50 L
|
○危険性・安全性リスクアセスメント計画の実施と 評価
○反応混合物の危険性
○反応生成物及び副産物の危険性・安全性
○スケールアップによる安全性・危険性の増大評価
○リスクアセスメントによる仕込み、反応、後処理等の安全対策、設備の評価
|
プロセス(工業化学)研究所、生産技術研究所
・有機合成化学者
・化学工学者
・分析化学者
|
|
パイロット製造
|
~500 L
|
○危険性・安全性対策の有効性と機能の検証と評価
○プロセス(標準操作法)の安定性と操作条件の制 御性
|
プロセス研究所、生産技術研究所(or工場、製造部)
・プロセス開発者
・製造現場作業担当者
|
|
PQ 製造
|
~5,000 L
|
○リスクアセスメントの結果と対策の教育訓練
○実製造でのプロセスの安定性・危険性・安全性の 対策評価
○スケールアップによる安全性・危険性の再評価と 対策
○商業生産を通じて安全性・危険性の再評価と対策
|
||
PV・商業生産
|
~10,000 L
|
工場(製造部)
・製造現場作業担当者
|
||



最後に、製薬メーカーは患者さんへ医薬品を安定供給する責任を、また、原薬メーカーは恒常的に品質規格に適合した原薬を安定供給する責任を負っています。そのため著者は「あなたが製造する原薬の医薬品を家族に飲ませられますか?」と製造前に製造担当者に口酸っぱく繰り返し言って来ました。それは「自分が製造する原薬に責任を持ちなさい」の気持ちを込めたものです。プロセス開発者も作成した標準操作法で製造現場が迷わず操作が出来、恒常的に高品質の原薬が得られ、製造作業者の健康、環境、危険性等も含め開発した製造プロセスに責任を持つ必要があると考えています。
更に、原薬のプロセス開発者・製造担当者は協力して原薬のアニュアルレポートをまとめ製造状況(標準操作法の操作条件と実際の製造条件の関係)と原薬の品質、不純物プロファイル、収率等との比較検討し、現行の標準製造法(条件範囲等)を更に最適化して向上させる努力を欠かさないことが重要であると考えています。ここまで、「医薬品原薬のプロセス開発に於ける実験室と製造現場の違い」についてお話を色々とお話してきましたが、実際にはコルベン(丸底フラスコ)で最適化した反応を含む操作条件でスケールアップ製造を実施しても殆どの場合問題なく期待した品質・収率で目的物を得ることができます。これは工程反応が少々の条件が外れても進行すること、反応条件下で原料・試薬・反応遷移状態中間体・生成物等の熱安定性と撹拌及び伝熱効果が十分に担保されていることが考えられます。


一般的に、医薬品原薬製造工程での固液分離のろ過で目的物を得る場合は遠心分離機を使用することが殆どですが、目的物が酸素・熱に不安定である場合は加圧ろ過乾燥機等を選択する必要があります。実験室でヌッチェ、製造現場で遠心分離機を使用する様に、実験室と製造現場でろ過機の原理が異なる場合が多々あります。例えば、目的物がろ過性の良い結晶では問題となりませんが、ろ過性が悪い場合はろ過に長時間を要するため、目的物の熱・酸素・湿気等の安定性を考慮してろ過機を選択する必要があります。また、ろ過性の悪い結晶は乾燥操作でも長時間を要することになります。ろ過性の悪い結晶何も良い結果をもたらしません。
ろ過操作のスケールアップでは、ろ過原理を合わせることと単位面積当たりの湿晶量(容量)をできるだけ一定にすることです。但し、ろ過性の良い湿晶の場合はこの限りではない。



コメント
コメントを投稿