7.プロセス開発で解決すべき課題について、
1)工程操作条件の最適化の課題(有機合成化学)
工程操作条件の最適化は、品質リスクマネージメントの一環として工程操作の品質リスク
アセスメントを実施し、データ取りの実験計画を作成し、計画に従いデータ取りを行い、
得られたデータから最適操作パラメータを設定(許容範囲と安全域を取った操作範囲と
操作目標値)することである。この時、最適操作パラメータの設定に当たっては、幾何
学的相似形実験機を用いたパラメータのデータ取りと操作したこと及び起こったイベン
ト全てを記載することが重要となる。また、スケールに合わせたスケールアップシミュ
レーションにより操作時間の予測、操作パラメータが製造実機の性能・能力から運転可
否の検証、示量的数値の予測計算、実機の性能・能力と原理の違いによる品質への影響
予測などを実施することである。更に、スケールダウンシミュレーションとして、スケ
ールアップシミュレーションで得られた最適化条件を基に、幾何学的相似形実験機を用
いて操作パラメータのワーストケース(操作範囲、操作予測時間など)で検証し、操作
状態の相似と品質への影響を確認することである。この様にして得られた最適化操作条
件で製造標準操作法を作成する。製造標準操作法に従い製造現場で操作範囲内の目標値
で運転することになるが、製造標準操作法で実機が運転可能か、実験機内の操作状態を
再現できるかが問題となる。このことからも、実験室、製造現場(パイロット製造及び
スケールアップ製造)では、反応槽内の運転状態の細かなチェックと全ての事象を詳細
なデータとして残すことは、プロセス開発だけでなく商業製造時の更なる最適化とスケ
ールアップ、収率の安定化、コストダウン、逸脱・失敗等の回避、製造機器の性能維持
並びに原薬の品質向上と安定化等々に有効手段となると考えている。実験室も製造現場
も工程操作の監視・観察が大切である。
図 36 に示す様に、品質リスクアセスメントの工程操作リスク(課題)の抽出は石川ダイ
アグラム等で全ての操作を書き出すことにより、クリティカルファクターの抽出・分
析・評価を容易とし、データ収集とクリティカルパラメータ用の実験計画を作成する上
で有効となる。
図36. 工程操作の石川ダイアグラム*

以上の観点から、プロセス開発に於ける有機合成化学の課題と問題点を挙げると表6の様
になると考えている。
表6. 「医薬品原薬製造プロセス開発に於けるスケールアップ」での有機合成化学の課題
と問題点
反応条件最適化解析法には、一変量解析と多変量解析があり、一変量解析が主流であっ
たが、現在では開発時間を短縮するために多変量解析が主流となりつつある。一変量解
析は、原料量と試薬並びに副原料比、滴下速度、触媒量、反応温度、反応時間等の操作
条件に関わるパラメータを一つずつ動かしデータを集め解析し、収率・純度・不純物プ
ロファイル・収率への影響から反応条件・操作条件の最適化を行う手法である。多変量
解析については、実際に実施したことがないためコメントできないが解析用のソフトが
販売されているので、興味のある方はソフトを確認して頂きたい。
有機合成化学の課題として実験室でのデータ取りを正確スピディーに効率良く行うため
には、遭えて一次スクリーニング(TLC)と二次スクリーニング(HPLC)に分け反応条
件等を絞り込む工夫も大切であると考えている(図 37)。急がば回れも必要。但し、デ
ータは出来るだけ数値化し分析と解析に耐えるデータ取りに心がける。反応追跡は
HPLCを用いて定量的に扱うのが一般的であるが、HPLC分析(二次スクリーニング)は
含まれる全ての物質を分析出来ている保証がない。TLC(一次スクリーニング)は定量
化が苦手であるが、反応液に含まれるすべての物質を一斉に分析でき可視化することが
出来るので、これらを組み合わることも大切である。実験室では、必要に応じて反応追
跡にTLC と HPLC を使い分け、反応条件の最適化時に HPLCを用いて純度、不純物量、
不純物プロファイル等を定量的に扱い数値で管理する。
図37. 実験室でのデータ取りの効率化(例えば、溶媒選択の一次スクリーニング)

例えば、HPLC 分析を反応追跡に用いる場合、1回の分析に 30~50 分間程度を要する。
また、分析用のサンプルの採取と調製に~10 分間程度要するとしたら、反応チェックの
判定に 40~60 分間程度掛ることになる。従って、反応チェックの結果は約 1 時間前の
反応状態を確認していることになる。例えば、図 38 に示す様に、反応が進行する場合
は、中間体或いは原薬を精製して品質規格に適合させるために反応液中の原料 <5.0%、
不純物 <3.0% でないと精製できないとしたら、本反応は 5~8 時間(この範囲であれば
ギリギリであるので、好ましくは 6~7 時間)で停止させる必要がある。本反応のHPLC
分析は 1 時間毎でなく、最低反応開始後 4 時間以降は30分毎にすべきである。従って、
最適な反応終点を得るためには、反応チェックの分析時間が10~20 分程度で終了する
HPLC 条件の採用、或いは反応条件の再検討による最適化が必要である。
図 38. 反応追跡に用いた HPLC 分析結果
2)スケールアップの課題(化学工学)
スケールアップは、実験室で得られた最適化データを基にスケールアップシミュレーシ
ョンを実施し、製造スケール・使用予定反応槽に合わせて変更が必要なパラメータを予
測計算して現場製造に適応させるパラメータに反映させることと考えている。特に、ス
ケールアップに於いては実験室と製造現場の反応槽との撹拌効率及び伝熱効率を相似
(同等化)させる計算、並びに延長する作業時間予測計算はスケールアップ後の原薬の
品質を確保するための重要なパラメータである(表7)。
表7. 「医薬品原薬製造プロセス開発に於けるスケールアップ」での化学工学の課題と問
題点
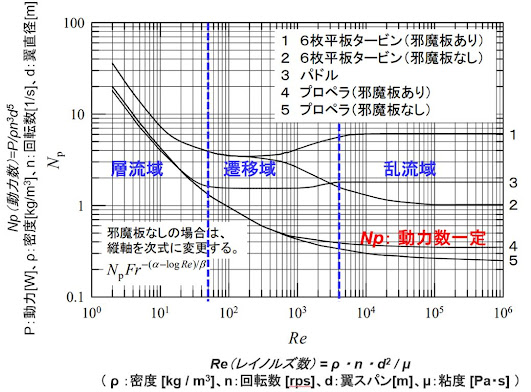
撹拌と伝熱(6.反応槽のスケールアップに必要な化学工学の予測計算)については既に
述べたが、撹拌は「単位体積当たりの撹拌動力」一定で、伝熱は「単位体積重量当たり
に加える熱量」一定でスケールアップする。このことから、スケールアップ製造に於い
ては化学工学の予測計算によりスケールに合わせた撹拌数及びジャケット内の熱・冷媒
温度への変更させる必要がある。
下記計算シート(図39)は、化学工学を知らない筆者が低温反応のスケールアップ時に
到達予測時間と冷媒温度の設定ができる様にお願いして、元大手製薬会社の化学工学の
専門家に作成して頂いた計算シートである。本ブログで計算シートを紹介させて貰って
いるので謝辞を伝えたい。この様な計算シートを作成しておくとパイロット製造から実
製造へのスケールアップ移行時に加熱(冷却)時間と大小の反応槽内の所定温度への到
達時間を相似させる熱媒・冷媒温度が予測できる。
本計算シートの使い方としては、
例えば、500 L 反応槽を用いて内容量 400 kg/Lを20℃ から -30℃ へ -50℃ の冷媒で冷却
する場合は約84分間を要しますが、同条件下で反応槽 5,000 L 反応槽を用いて内容量
4,000 kg/Lと10 倍のスケールアップの場合に約 183 分間(約2.2 倍の時間)を要する
(図 39)。これは反応槽のサイズが 10 倍に上がっても伝熱面積が 4.6 倍にしか増加し
ていないためである。同じ時間で冷却するためには、冷媒温度を -110℃ 程度にする必要
があると計算される。この計算結果から、発熱を伴う低温反応のスケールアップに於い
ては、スケールアップ前後で同等の反応槽内温度を保つために冷媒温度を下げる、試
薬・副原料等の滴下速度を遅らせる、撹拌速度を上げる等を考慮する必要がある。スケ
ールアップ後の冷媒温度が計算予測できれば反応条件、製造設備・機器等の選択に役立
つと考えている。
図 39. 同一冷媒温度によるスケールアップでの冷却時間<到達予測時間(θ)計算
シート7)>
3) 乾燥操作の課題
原薬・中間体の乾燥に用いられる乾燥機の種類(原理)には、棚式乾燥機、コニカル乾
燥機、振動乾燥機、ろ過乾燥機等々がある(図 40)。乾燥条件の最適化は、先ず、湿
晶の状態・性質(酸化され易いか?、熱安定性は?、wet率は?、結晶サイズは?等々)
を理解し、それぞれの乾燥機の原理(静置・振動・回転・撹拌、減圧・送風など)から
乾燥機を選択する。次に、乾燥温度、減圧下か、常圧か、減圧度を設定する。但し、真
空乾燥機の場合は、減圧度と排気量は工場に設置されているの真空ポンプの能力に依存
するので、工場のポンプの能力を加味しながら実験室でデータ取り計画を立てる必要が
ある。しかしながら、乾燥温度は熱安定性から設定されるが、減圧度はほとんど考慮さ
れていないのが現状だと思う。乾燥温度は原薬・中間体の熱安定性と望む乾燥時間(一
夜、8時間以内)で設定されることが多い。
図 40. 医薬品原薬のプロセス開発から製造時に用いられる一般的な乾燥機
また、医薬品原薬の乾燥工程条件を設定するためには、晶析後にどの様な結晶状態(溶
媒和物・水和物或いは無水物)で得られるか、製品である原薬がどの様な結晶形態(無
水物或いは水和物)・結晶多形であるかを理解しておく必要がある。結晶多形は熱、溶
媒媒介及び圧力などにより転移を起こすことがあることから、乾燥時の熱、圧力などに
注意を払う必要がある。
最終原薬・中間体の乾燥に用いられる減圧乾燥の原理は、どの乾燥機でも真空ポンプを
用いて減圧下とし結晶中に含まれる或いは付着した溶媒・水の沸点を下げある一定温度
で加熱し水等を気化させポンプの排気能力(量)を利用して水分等を強制的に庫外へ排
出させることです。スケールアップ時の乾燥機の選定は湿晶の性質の理解から始める
が、湿晶の結晶サイズ並びにwet率などを考慮して行う。棚式真空乾燥機は結晶サイ
ズの大小及び wet率の高い湿晶であっても乾燥することが出来る。しかしながら、
wet 率の高い湿晶の場合は塊となることが多く時々乾燥を中断して結晶を解す操作が
必要となる場合がある。コニカル真空乾燥機の場合は、結晶サイズが大きく、wet 率
が低い時は問題ないが、結晶サイズが小さく、wet 率が高い湿晶はコニカルの中で塊
まりとなって乾燥されることが多く、大きく硬い塊はコニカルのGL 面を損傷させる
或いは排出できない場合がある。このことから、実験室で相似形コニカル容器を用い
て回転条件を検討する必要とする(図 41)。しかしながら、コニカル乾燥機は回転
することから結晶(原薬)の均一性が確保できるメリットがある。
図41. コニカル乾燥機の実験室から製造現場へのスケールアップ検討a)
結晶形に関する通知:薬食審査発0616第1号
既承認医薬品の原薬を異なる結晶形の原薬に切り替える場合又は既承認医薬品の原薬と
異なる結晶形の原薬を追加する場合には、原則として、承認事項一部変更承認申請で取
扱い、異なる水和物/無水物の原薬に切り替える場合には、原則として、代替新規承認
申請で取扱うこととする。
小山らはb)、湿晶エリスロマイシン誘導体の乾燥を50℃ の通気乾燥、60℃での真空乾燥
及び 25℃ + 60℃ の真空乾燥で乾燥時間を比較している。 60℃ での真空乾燥すること
により 7 時間で残留溶媒の規格適合品を得ている。一時乾燥を 25℃ で 4 時間真空乾燥
後に 60℃ に昇温し追加真空乾燥(6時間)を実施することにより 60℃ のみの時より約
1 時間速く残留溶媒規格適合品を与えることを示している(図 42)。乾燥条件の最適化
は原薬の熱安定性・乾燥時間等の点かも重要である。
図 42. エリスロマイシン誘導体の乾燥条件による残留溶媒量b)
この様に、晶析で得られる湿晶は水和物或いは溶媒和物の状態で得られることが多く、
湿晶の乾燥温度(乾燥原薬の熱安定性(温度 X 時間)から決定することになる。しかし (水和量)が異なることから、結晶多形でなく疑似多形と呼ばれている。
図 43. 医薬品原薬カルバゾクロムスルホン酸ナトリウム水和物と無水物の結晶格子c)
式真空乾燥機のスケールアップ(一次乾燥)例
棚式真空乾燥機は殆どの性状・性質の湿晶も大量に乾燥でき、凍結乾燥(水溶性原薬、
ペプチド或いはたんぱく質水溶液など)にも用いられる。棚式真空乾燥機は湿晶をトレ
イに分け載せて乾燥するため、結晶が微細でwet率の高い湿晶では途中で解さないと硬い
塊となることがあり均一性にも難があるため庫内の乾燥晶の均一性試験が必要となる。
棚式真空乾燥機のスケールアップで最も気を付けるべきファクターはトレイに載せる湿
晶の厚みである。この湿晶の厚みが異なると棚板から伝わる熱量が単位体積当たり一定
にならない。棚式乾燥機でのスケールアップでは、真空(減圧)度は製造現場の真空ポ
ンプの性能に依存することから設定せず、温度は棚温度を一定温度の温水を流し管理す
るためトレイの大小に係わらず湿晶の厚みを同一に広げ「単位体積当たりにかかる熱量
一定」を守って実施する(図 44)。この様にして乾燥のスケールアップを実施するとほ
ぼ同等の乾燥時間で乾燥できる。更に、棚式真空乾燥機の機器本体で重要なことは、棚
は温水が通り加熱するジャケットの役目を持っているため、如何に棚板全体に、棚板間
に差がなく均一に温水が流れ一定温度を保ち、棚板とトレイの間に隙間が出来ない真っ
平になっていることです。これが乾燥機製作会社のノウハウであり、棚式乾燥機を購入
するうえで重要な見極めと聞いている。トレイも同様である・
図 44. 棚式真空乾燥機のスケールアップ例

水和物原薬の二次乾燥(調湿)
水和物原薬の乾燥操作は困難となることがある。湿晶が溶媒和物の場合は、所望の水和
物量原薬を得るために一次乾燥で先ず無水物へ導き、次に水和物原薬を作製する二次乾
燥を一定の相対湿度に調整した所定温度の加湿不活性ガス(窒素など)を通気させるこ
とにより調湿する。湿晶が水和物の場合は乾燥温度を最適化し一時乾燥で直接所望の水
和物量の原薬を得るか、湿晶に一定の相対湿度に調整した所定温度の加湿不活性ガスを
通気させることにより所望の水和物量の原薬を得ることになる。
例えば、植草らc)によれば、セファレキシン(セフェム系抗生物質)は無水物が原薬であ
るが、原薬に相対湿度と温度条件(温度は不明)により無水物(R.H.=相対湿度 0%)か
ら 2.5 水和物(R.H. 100%)の5段階の水和物を与えることが知られている(図 45)。こ
の様に、調整した加湿不活性ガスを乾燥結晶に通気させると相対湿度に合わせて結晶内
に水の取り込み或いは吐き出しが起こり所望の水和物量の原薬を得ることが出来る。
図 45. セファレキシンの水蒸気吸着測定c)
4)スケールアップシミュレーションによる変動要因(操作条件)のデータ取り(パラメ
ータの設定)
有機合成化学者は、プロセス開発時の操作パラメータの取得するために類似の反応を
Reaxys、ChemFinder等の検索により見出し、その条件を用いて実際の化合物に適用さ
せるのが一般出来である。その結果を基に、化学工学が協働して必要なデータ取りを
実施する(表8)。しかしながら、ジェネリック或いは新薬メーカーからの原薬の GMP
製造依頼では、既に工程操作条件が指定されていることが多く、自社の製造実機に合わ
せたプロセス開発となる。どちらにしても、スケールアップに於いては、実験室で得た
最適操作パラメータからスケールアップシミュレーションとして品質リスクマネージメントとして品質リスクアセスメントを実施することになる。その結果を基に必要な或いは足りない実験計画の策定と計画に合わせたデータ取りを実施する。得られで操作パラメータは安全域を持たせ範囲に操作目標値を指定し、特にクリティカルパラメータは目標値を外れない様に注意を則す必要がある。最適化された製造操作法の操作パラメータ、操作時間などを含め製造現場を再現させるために幾何学的相似形実験機を用いてスケールダウンシミュレーションにより検証・実証する。この時、ワーストケースとして操作範囲の上限値或いは下限値により操作し、品質への影響を検証しておくことを勧める。
表 8. スケールアップに必要なデータ
実験室から製造現場へのスケールアップでの注意点については既に述べてきたが、まと
めると表 9 になると考えている。原材料の品質では、特に出発物質の品質が原薬品質に
重大な影響を与えることがあるので、製造業者・製造場所・製造法の変更時は要注意で
ある。反応に於いては、実験室の撹拌(流動)状態、伝熱状態と操作時間などを製造現
場で全て相似させる必要がある。このことから、実験室でのスケールアップ時に掛る操
作時間の予測とそれに余裕を持たせた時間での操作溶液中の原料・反応中間体・生成物
(目的物)・原薬等の熱安定性が担保(安全域を確保して)されていなければならな
い。最後は、人が製造設備を運転・工程操作などを実行するため、製造標準操作法の手
順に従い誰が運転・操作しても間違いなく同じ運転状況が得られるように記載すべきで
ある。このことから、ヒューマンエラーを無くすために自動化が進んでいるが、これも
プログラムの書き込みも人の手で行うのでヒューマンエラーに注意を要する。スケール
アップ時の留意点に注意を払い実験室(有機合成化学)のデータを基に現場製造へスケ
ールアップさせるためには、実験室と製造現場の相違点を理解し「スケールアップシミ
ュレーション」を実施する。本シミュレーションで得られたスケールに合わせた操作条
件を基に使用予定の製造現場機器を選定し、その幾何学的相似形装置を用いてスケール
アップシミュレーションで得られた操作条件(操作時間を合わせ)による「スケールダ
ウンシミュレーション」で検証すべきです。また、スケールダウンシミュレーション
は、操作範囲の上限値或いは下限値での品質への影響も検証しておくべきです。
表9. スケールアップに係る留意点
*操作時間は、操作範囲内でも少しの条件の違いにより差が生じるため、一般的に設定しない。
得られたデータからスケールアップへの品質リスクマネージメントの実施と現場設備・
機器からのスケールダウンシミュレーション(幾何学的相似形装置によるデータ取り)
による検証する。この時、スケールアップ時に予測される全てのパラメータをスケール
及び時間軸で予測計算し、その計算値に十分な安全率を掛けワーストケースでのスケー
ルダウンシミュレーションで設定パラメータの検証を行う必要がある(図46)。スケー
ルアップ製造は、有機合成・化学工学・分析化学・製造現場・品質保証の担当者の協働
により必要なデータの取得と解析並びに製造設備・機器の能力から品質リスクアセスメ
ントを実施し、スケールアップシミュレーション並びにスケールダウンシミュレーショ
ンを実施し目標品質(純度・不純物プロファイル・不純物量・収率等)が達成される結
果が得られれば、表10 に示す様な「今までのスケールアップ倍率の考え方」でなく、一
気に100~1000倍のスケールアップ製造が可能となると考えている。
図46. スケールアップ・スケールダウンシミュレーションの設備と実施方法

今までのスケールアップは、表 10 に示す様に、経験上 1~2 L から 10~20 L、更にパイ
ロット製造として50 L~100 L へ、更に 500~1000 Lへと 10倍毎にスケールアップすべ
きと考えられて来た。この考え方は、失敗を最小限に留めること、スケールアップは難
しいとの考え、最適化操作条件の適応範囲を確認していないこと、化学工学的(スケー
ルアップ・ダウンシミュレーション、幾何学的相似形実験機、予測計算など)な思考が
なされてこなかったこと、10 倍のスケールアップ前後での操作の違いが少なかったこ
と、或いは操作時間等の差異により得られる中間体・原薬の品質・収率を比較検証し製
造の標準操作法に反映させながらスケールアップ製造に成功していたこと等から来てい
ると考えられる。しかしながら、今まで説明して来た様に幾つかの相似を達成させるこ
とにより、一気に100~1,000 倍へのスケールアップも夢でなくなり、プロセス開発、標
準(製造)操作法の開発期間を短縮でき、コスト削減にもつながる。医薬品原薬のプロ
セス開発に於けるスケールアップでは、化学工学的思考を取り入れ、実験機と製造実機
の違いを理解、スケールアップシミュレーションの実施、品質リスクマネージメントの
実施(品質リスクアセスメントとして実験計画と検証)、スケールアップ予測計算(撹
拌、伝熱、操作時間など)、熱安定性の確保、操作条件に安全域を取った範囲に実際の
操作目標値の設定、操作範囲のワーストケースでの原薬の品質担保、スケールダウンシ
ミュレーション検証などが重要になると考えている。
表10. 今までのスケールアップ倍率の考え方

5)操作パラメータの設定
反応条件の最適化は、原料、試薬、副原料、触媒及び反応溶媒の品質と量比、投入速
度、温度、濃度、圧、光並びに触媒或いは不均一試薬(触媒を含む)の表面積等のパラ
メータがどの様に中間体及び原薬の品質(純度、不純物プロファイル及びその推移)に
影響を与えるか、更には操作時間と反応中間体・生成物の熱安定性等、操作ファクター
を含め設定する必要がる。
筆者は製薬会社へ就社した当時に反応条件の最適化に於いて一度に二つの因子を同時に
変換するとどの組み合わせが最適なのか分からなくなると言われてきた。実際、このこ
とから反応条件因子(ファクター)を一変量変換で最適化し、条件因子を範囲(デザイ
ンスペース?)で設定してきたがスケールアップで大きな問題は生じなかった。
例えば、F. Huangらは、Phytosterol
のエステル化で一変量変換(反応時間、反応温度、
PUFA(polyunsaturated fatty acid)とphytosterol比、及び触媒量)の最適化例を示して
いる(図 47)。一変量変換で得られた反応最適因子を組み合わせて反応条件を最適化し
ている。
図 47. 一(単)変量変換による反応条件の最適化実験例8)
得られた全ての反応操作条件パラメータに上限値・下限値を求め、中間体・原薬に与え
る収率・純度・不純物プロファイル(品質規格)の影響を考慮して、これらのパラメー
タに設定範囲(操作範囲)を設定することである。特に、重要パラメータの設定では、
操作をより安定的に、より恒常的に原薬の収率・純度・不純物プロファイル(品質規
格)を保証するために操作範囲を狭めるが安全域を確保して設定することを薦める。ま
た、製造現場では目標(設定)値(1点)を操作値の目標としてより狭い範囲で製造管理
することが、恒常的に原薬等の安定した品質規格の適合と収率の安定につながる(図
48)。この安全域を設けることがPQ (Process Qualification) 或いはPV
(Process
Validation) の成功へ導く重要な鍵となると考える。
図48. 操作パラメータの設定範囲の考え方

例えば、得られた最適値の反応(操作)温度は、熱安定性のデータ並びに反応速度・収
率・不純物量・不純物プロファイルから許容範囲を設け、この範囲に最低10℃以上の安
全閾を設けて設定(操作)範囲をする。更に、不純物量、不純物プロファイル並びに収
率等から設定範囲内の最適値(≒中央値)を目標(設定)値として設定する(図 46)。
副原料、試薬、触媒並びに酸塩基量等の量比は原料の残量、不純物量、不純物プロファ
イル、収率或いはその組合せ等により、同様に操作許容範囲(安全域=操作許容範囲-
操作設定範囲)→操作設定範囲→操作目標値を設定する。GMP 製造では、基本的に操作
範囲でパラメータを管理するが、操作目標値での管理をすることにより製造現場での製
造結果を基に品質・収率のバラツキから操作・設備の何処に問題があるかが把握出来
る。また、商業生産に於いても操作目標値での管理することにより、医薬品原薬の品質
と収率のバラツキを製造毎と年間製造報告書(アニュアルレポート)での検証比較が容
易となり恒常的に品質規格範囲上限の高品質で中間体・原薬を与える目標値と操作範囲
を狭めることができ、更なる最適な目標値を設定し直すことが可能となる。
6)製造法操作法の設定品質リスクマネージメントとしての品質リスクアセスメント
プロセス開発に於ける操作法の品質リスクアセスメントはプロセス開発に於けるスケー
ルアップ時の工程操作にどの様なリスクが潜んでいるかを検証するために大切である。
品質リスクアセスメントはスケールアップ時のどの操作にどの様なデータが必要か、そ
のデータ取りをどの様に進めるか、或いは既知の科学的データをどの様に調査するかを
判断するものです(表11)。プロセス開発者は、図34に示す工程操作のフィッシュボー
ンなどにより工程の全ての操作を抽出し、工程操作に潜むで品質に重大な影響を与える
プロセスファクターを見つけ出し、その中で品質に重大な影響を与えるクリティカルパ
ラメータを詳細に設定する必要がある。そのためには、品質マネージメントとして品質
リスクアセスメントの実施と実験計画を策定し、この計画に従いデータ取りを実施し、
並びにデータ解析によるクリティカルファクターの抽出とそのパラメータの設定を実施
すべきである。品質リスクアセスメントと実験計画例を下記に示した(表 11、12)。
表11. 工程の品質リスクアセスメント例

7)サクラミル原薬9)の実生産スケールに於ける標準的製造方法
サクラミル原薬S2モックに記載されている Step 1 の製造法を下記に示しているが、操作
パラメータは1点の設定値のみが記載されている(工程「サクラミル原薬承認申請書(或
いは、MF)記載製造法例」)。しかしながら、操作パラメータが1点では製造現場の作
業が困難でのあり、逸脱してします。製品標準書の製造法には製造現場の作業が容易に
出きる様に操作パラメータを範囲で記載されている(「製品標準書製造方法記載
例」)。この操作範囲の上限値或は下限値で操作した時、中間体或は原薬の品質を保証
するためには操作範囲に安全域を設けた許容範囲を設けることを推奨する。全ての操作
パラメータの値は実験データと品質規格から科学的に設定する必要がある。設定(目
標)値、操作範囲及び許容範囲の設定の考え方を図 46 に示した。
Step 1

<サクラミル原薬承認申請書(或いは、MF)記載の製造法例>
Step 1で得られたCP-7[2] 『(250 kg)』注1)、3,5-ビストリフルオロメチルベンジル
ブロマイド (CP-8)『(215 kg)』注1)及びジクロロメタン『(750 L)』注1)を仕込
み、テトラブチルアンモニウムブロミド『(50 kg)』注1)及び“50%” 注3)水酸化ナ
トリウム水溶液『(750 L)』注1)を内温20℃で加えてはげしく 6 時間かき混ぜる。ジ
クロロメタン及び水を加え、分液し、有機相を希塩酸で洗浄する。有機相を濃縮し、エ
タノールの質量に対して20~35%注4)の質量に相当する水を加えた後、《毎分0.15~
0.5℃》注4)で『18℃』注2)まで冷却し、かき混ぜる。結晶を分離し、エタールで洗
浄する。結晶を『42.5℃』注2)で乾燥してエチル (2R,4S)-4-{[3,5-ビス(トリフルオロメ
チル)ベンジル](メトキシカルボニル)アミノ}-2-プロピル-6-(トリフルオロメトキシ)-3,4-ジ
ヒドロキノリン-1(2H)-カルボキシレート[3](サクラミル)を得る。(収量 360 kg,収率
90%)
<製品標準書製造方法の記載例>
Step 1で得られたCP-7と3,5-ビストリフルオロメチルベンジルブロマイド(CP-7の1モル
に対して1.0~1.1モル当量)をジクロロメタン(CP-7の1kgに対して2~4 L)中で混合す
る。温度を12~25℃に保ちながら、テトラブチルアンモニウムブロミド(CP-7の1kgに
対して0.1~1.0 kg)と水酸化ナトリウム水溶液(CP-7 の1kgに対して47~50%溶液で2
~4 L)を加える。反応が終了すれば、ジクロロメタンと水を加え、分液し、有機層を希
塩酸で洗浄する。有機層を濃縮し、蒸留しながらエタノールに溶媒置換する(終濃度は
CP-9 の1kgに対して4.5 Lにする)。水(エタノールの質量に対して25~35%の質量)を
加え、14~26℃で攪拌する。固体をろ過し、エタノールで洗浄し、50℃以下で乾燥して
CP-9 (サクラミル)を得る。
上記の様に、「原薬承認申請書(或いは、MF)製造法の記載例」と「製品標準書製造方
法の記載例」は異なり、製品標準書製造方法の記載例には各操作のパラメータを操作範
囲で記載している(表13)。これは実際の操作パラメータが操作範囲内のブレであれば
品質規格に適合する中間体或いは原薬を与えることを保証しており、範囲内のブレを最
小限にするためには目標値を設けることである。また、操作範囲のパラメータを確実に
保証するためには、操作範囲の上下に安全域を設けた許容範囲を確保することである。
この安全域の広さがパラメータのリスクの程度を判断する材料にもなる。従って、きめ
細かな幅広いデータ取りが、より確実な製造操作法を作り上げる基となる。
表 13. 反応条件の設定値(最適値)、操作範囲値及び許容範囲値の設定例

但し、PMDAは製造承認後に承認申請書(或いは、MF)に記載した 1.2倍以上のスケー
ルアップ製造は認めていない。操作(反応を含む)時間は一般的に設定しないが、操作
時間は種々の不具合などにより延長する可能性が非常に高いため可能な限り長く熱安定
性のデータを取り品質への影響を検証して最大操作時間を確定しておくことを薦める。
不具合が発生しても操作溶液中の品質確認と最大操作時間内であれば中間体・原薬の品
質が担保できていることをPMDA或いは委託先へ説明できる。
8)より進んだ方法(多変量解析)によるデータ取りの実験計画
今までのデータ取りはパラメータの1項目のみの一変量解析を行っていたが、より進んだ
方法として同時に複数の操作条件パラメータを変更しデータ取りを行う多変量解析によ
り最適なパラメータを求める方法が提案されている。これについては実施したこともな
く、理解もしていない。
最近の反応操作条件の最適化は多変量同時変換が主流になりつつあり、L. Sanchez10) は
多変量解析実験法について述べている(表 14) 。一変量変換では因子数が増えると実験
数が膨大となるが、反応条件因子(1. 反応温度、2. 濃度及び3. 触媒種類)をそれぞれの
因子を二変量(例えば、反応温度 120℃(-1)と 160℃(+1)に)させその組み合わせ
実験により 23の実験数で収率に与える影響を確定している。詳細は割愛するので資料
12月4日まで存在確認済み)。その中で反応温度が最も重要な因子であり、副原料量は少
ない方がよく、塩基量は多い方がよかったと読み取れ、更なる細かな各因子の変量組み
合わせデータ取りが必要となると思うが、得られたデータを多変量変換解析法により最
適パラメータと収率を予測している(図 49)。これについては、多変量変換解析法のソ
フトが市販されているので興味ある方は参考にされたい。
図 49. 多変量解析の変更項目とパラメータ、並び実施結果10)
9) 医薬品原薬のプロセス開発に於けるスケールアップ
医薬品の開発確率は、0.003~0.005% 程度(2018年度)であり、医薬品原薬のプロセス
開発を効率的に進める必要がある。原薬のプロセス開発は、GMP製造の初期から商業製
造法を完成させるのでなく、開発ステージに合わせた製造スケールに合わせた標準操作
法・品質目標(原薬品質が毒性試験で担保されている範囲内)を定めて堅牢性・恒常性
を順次高めて行くべきである(図50)。開発ステージによるプロセス開発の大まかな内
容を記載する。
図50. 治験薬の開発ステージに合わせたプロセス開発11)

製造標準操作法の確立於いては、① 既に製造法が存在する場合はプロセス開発で既存製
造法に従いトレース実験と自社の製造設備・機器の特性・能力などを把握しスケールダ
ウンシミュレーションにより操作パラメータの検証と自社に合った製造操作条件の確
立、並びにスケールアップシミュレーションによる予測計算と熱安定性などを担保した
製造スケールに耐える製造法(商業製造法)を開発する。② 新規合成ルートでのプロセ
ス開発では、プロセス開発は共通となるが、出発物質の選定、合成ルートの最適化とそ
の品質規格・試験方法など新規の確立が必要となる。しかし、新規合成ルートが確立で
きればコスト及び品質の点で優位になる可能性がある。
新薬のプロセス開発
・開発前期 (前臨床(Non-GMP)~Phase I(GMP)): ラボスケールで設定した最
適操作(製造)条件をスケールアップシミュレーションによりパイロットスケールに
合わせて再設定し、パイロット製造を実施(スケール:~50 kg)し、検証する。
・開発中期(Phase I~II): パイロット製造の評価とデータからスケールダウンシミュ
レーションの実施と実験室で必要なデータ取りと再最適化⇒必要に応じてクリティカ
ルパラメータの再設定 ⇒ GMP製造(スケール:~100 kg)。スケールに合わせた操作
条件のスケールアップシミュレーションとスケールに合わせた操作時間の予測と熱安
定性の確認、撹拌状態の相似させる撹拌数の予測計算並びに伝熱状態を相似させる冷
媒・熱媒温度の予測計算を実施する。スケールアップ条件を基に幾何学的相似形実験
機を用いたスケールダウンシミュレーションによる最適化操作条件の検証と必要に応
じて再設定。GMP製造による製造標準操作法の堅牢性確認と操作条件(パラメータ)
及び品質のバラツキ検証と品質リスクアセスメントの実施。
・開発後期(Phase IIb~III): GMP
製造法の再評価と製造バッチを重ね製造標準操作
法の堅牢性と安定性の向上⇒商業生産(実生産(プロセス:~1,000kg)へ向け製造法
の堅牢性と品質の安定性並びにコストダウン(回収等を含め))へ向け科学的・合理
性に基づきクリティカルパラメータの決定と管理、操作条件の最終最適化する。プロ
セス開発の最終章として製造設備・機器の設定として PQ: Process
Qualification、製造
法の堅牢性・安定性の確認としてPV: Process
Validation により検証・実証する。
・商業生産 : 商業生産の製造標準操作法は PVの妥当性が実証できれば決定 ⇒ 製造承認
申請書(後発品では、MF:Master File)の製造に関する項目を作成 ⇒ 製造販売承認
申請書提出 ⇒ 製造承認後、実生産(プロセス:~1,000kg)で製造バッチを重ね製造
結果(アニュアルレポート等)から製造法及び品質規格等の妥当性を再検証し、更な
るコスト低減と最適化を実施する。後発品(ジェネリック)対策として、医薬品原薬
のコストダウン並びに品質等の向上に向け製造データの解析と実験データの取得を重
ね PVを実施する。製造販売承認申請の一部変更(一変)承認申請(或いは、MF の場
合も一部変更(一変)承認申請)して標準操作法を最新製造法へ更新し、他社・後発
品メーカーの追随を許さないためにも参入障壁を向上させる絶え間ない努力が必要で
ある(図 48)。
10) 反応、溶媒、原料・試薬・中間体の作業者への危険性、安全性(毒性含む)、環境
等への負荷
グリーンケミストリーが叫ばれる中、医薬品原薬のプロセス開発に於いても作業者の健
康・安全性に配慮し、環境への負荷及び危険性を最小限にする必要がある。製造量が増
えると使用する溶媒、原料・試薬等は増加し、危険性(発火、爆発等)、安全性(変異
原性等の毒性)、廃棄物量が比例して増大する。このことから、プロセス開発する上で
も増大する溶媒、高価な触媒、金属等の回収、反応効率の最大化、製造現場での作業
(者)の安全化を目指す必要がある。
反応等の溶媒の選択では、
メガファーマであるサノフィ―12)は、ICH のガイドライン、健康への影響、環境負荷、
廃棄・リサイクル、爆発性、反応性・安定性等をシートにまとめ、製造溶媒の選択基準
として発表している(表 14)
表 14. Sanofi の溶媒の選択基準

反応溶媒の選択基準
Sanofi13)
は、表 15 に示す様に、反応溶媒の選択基準もまとめている。反応溶媒(晶析
溶媒)の選択は、SciFinderなどの検索から類似の反応例を基に反応溶媒を選択している
のが一般的である。類似構造化合物の反応例から溶媒を選択し、実験室で実化合物の反
応へ適用して反応性、生成物の品質(副反応・純度、不純物量及び不純物プロファイ
ル)、収率等を確認して決定する(図35. 実験室でのデータ取りの効率化(例えば、溶
媒選択の一次スクリーニング)に示した様に)。また、反応・抽出・晶析なとの溶媒は
溶解度、電気陰性度、沸点、反応性、反応効率、反応時間、不純物生成量、不純物プロ
ファイル、副反応、冷却効率、後処理、水溶性或いは非水溶性溶媒、有害性、経済性
(コストと回収のし易さなど)等も考慮すべきであると考えている(図 14)。特に、最
終精製工程に用いる晶析溶媒は溶解度、精製度、晶析効率、晶析率並びに ICH 残留溶媒
のガイドラインなどに考慮して選定すべきである。次に、反応・晶析・抽出等の溶液濃
度は製造効率・原薬コストに直接影響を与える。このことから、反応濃度は 1 mol/L を中
心に反応様式(分子内或いは分子間)、副反応、反応性並びに流動性(撹拌性)などを
考慮して設定する。抽出溶媒は抽出効率・抽出時間(完全分離時間を含む)を考慮する
ため、目的物の溶解度、溶媒との反応性、エマルジョンの有無、安全性・危険性等を考
慮して設定すべきと考えている。抽出溶媒量は目的物の3~5 V (W)/W の範囲で、晶析溶
媒量は精製度・晶析効率を考慮して目的物の 4~8 V (W)/W の範囲で設定するのが望まし
いと考えている。
表 15. Sanofiの反応溶媒の採用基準

例:反応溶媒の違いにより生成物が異なる。
塩化チオニル*を反応剤と溶媒を兼ねると定量的に化合物 2
を与えるが、反応溶媒を変更
しエーテル系である MTBE**
を用いると化合物 3
を経由して同様に化合物 2
を与える。
また、塩化メチレン**を用いると化合物 6
を与える。この様に、反応溶媒は、同じ試薬
を用いても溶媒が異なると異なった生成物を与えることから、目的物を高品質で、高収
率で与え、副生物が少ない或いは生成する不純物が除去し易いことも優先して選択する
ことが大切と考える。






























































コメント
コメントを投稿